CD-HIT User’s Guide
November, 2006
http://bioinformatics.org/cd-hit/
Program written by Weizhong Li
1. Introduction
2. Algorithm
2.1. cd-hit clustering algorithm
2.2. algorithm limitations
2.3. cd-hit-2d comparing algorithm
2.4. DNA/RNA clustering & comparing
2.5. psi-cd-hit algorithm
3. User’s Guide
3.1. installation
3.2. cd-hit
3.3. cd-hit-2d
3.4. cd-hit-est
3.5. cd-hit-est-2d
3.6. cd-hit-para.pl
3.7. psi-cd-hit.pl
3.8. psi-cd-hit-2d.pl
3.9. incremental clustering
3.10. hierarchically clustering
4. CD-HIT tools
4.1. plot_len.pl
4.2. clstr_sort.pl
4.3. clstr_merge.pl
4.4. clstr_renumber.pl
4.5. clstr_rev.pl
5. FAQ
6. References
Introduction
CD-HIT was originally a protein clustering program. The main advantage of this program is its ultra-fast speed. It can be hundreds of times faster than other clustering programs, for example, BLASTCLUST. Therefore it can handle very large databases, like NR.
The 1st version of this program, CD-HI, was published and released in 2001. The 2nd version, called CD-HIT, was published in 2002 with significant improvements. Since 2004, CD-HIT has been hosted at bioinformatics.org as an open source project.
Since its release, CD-HIT has been getting more and more popular. It has a significant user base, I estimated at over 1000 users. It is used at many research and educational institutions. For example, at UniProt, CD-HIT is used to generate the UniRef reference data sets (http://www.pir.uniprot.org/database/DBDescription.shtml). It is also used in PDB to treat redundant sequences (http://rutgers.rcsb.org/pdb/redundancy.html).
Since 2005, I started the further development of CD-HIT, added more features to existing programs and wrote several new applications. Currently, I have 8 programs in the CD-HIT package: cd-hit, mcd-hit, cd-hit-2d, cd-hit-est, cd-hit-est-2d, cd-hit-para, psi-cd-hit, psi-cd-hit-2d. I also developed some utility tools, written in Perl, to help run and analyze CD-HIT jobs.
Current CD-HIT package can perform various jobs like clustering a protein database, clustering a DNA/RNA database, comparing two databases (protein or DNA/RNA), generating protein families, and many others.
This program is still under active development, new features and new programs will be out in the future.
Algorithm
Algorithms for CD-HI and CD-HIT were described in three papers published in Bioinformatics.
1. Clustering of highly homologous sequences to reduce the size of large protein databases. Weizhong Li, Lukasz Jaroszewski & Adam Godzik. Bioinformatics (2001) 17:282-283, PDF, Pubmed
2. Tolerating some redundancy significantly speeds up clustering of large protein databases. Weizhong Li, Lukasz Jaroszewski & Adam Godzik. Bioinformatics (2002) 18: 77-82, PDF, Pubmed
3. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Weizhong Li & Adam Godzik. Bioinformatics (2006) 22:1658-1659 PDF, Pubmed
I suggest that you read these papers if (1) you want to understand more details about the algorithm or (2) you want know why it is so fast. If you don’t have time to read these papers, the algorithms are summarized below.
CD-HIT clustering algorithm
Clustering a sequence database requires all-by-all comparisons; therefore it is very time-consuming. Many methods use BLAST to compute the all vs. all similarities. It is very difficult for these methods to cluster large databases. While CD-HIT can avoid many pairwise sequence alignments with a short word filter I developed.
In CD-HIT, I use greedy incremental clustering algorithm method. Briefly, sequences are first sorted in order of decreasing length. The longest one becomes the representative of the first cluster. Then, each remaining sequence is compared to the representatives of existing clusters. If the similarity with any representative is above a given threshold, it is grouped into that cluster. Otherwise, a new cluster is defined with that sequence as the representative.
Here is how the short word filter works. Two proteins with a certain sequence identity must have at least a specific number of identical dipeptides, tripeptides and etc. For example, for two sequences to have 85% identity over a 100-residue window they have to have at least 70 identical dipeptides, 55 identical tripeptides, and 25 identical pentapeptides. By understanding the short word requirement, CD-HIT skips most pairwise alignments because it knows that the similarity of two sequences is below certain threshold by simple word counting.
Another reason why CD-HIT is so fast is the use of an index table. I just use very short word with size 2~5. For instance, the total number of possible pentapeptides is only 215 (each position has 21 possibilities, 20 amino acids plus “X”), and the index table requires only 4 million entries, which just matches the RAM scale of current computers. Index table makes the counting of short word very efficiently. And a longer word is more efficient than a shorter one.
Algorithm limitations
A limitation of short word filter is that it can not be used below certain clustering thresholds. In a worst case scenario (figure below), when mismatches are evenly distributed along the alignment, the numbers of common short words are minimal. So theoretically, pentapeptide, tetrapeptide, tripeptide and dipeptide could only be used for thresholds above 80%, 75%, 66.67% and 50% respectively.
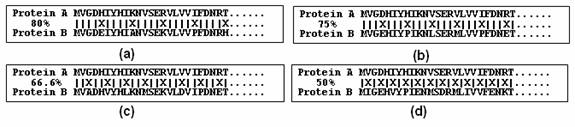
Short word filtering is limited to certain clustering thresholds. Evenly distributed mismatches are shown in alignments with 80%, 75%, 66.67% and 50% sequence identities. The number of common pentapeptides in (a), tetrapeptides in (b), tripeptides in (c), and dipeptides in (d) can be zero.
However, biological sequences are not lines of random letters; proteins usually have more conserved regions and more diverse regions as the result of specific constraints of evolution. Situations such as in above figure are very rare in the real world, and the actual number of common short words is much higher than in the worst case scenarios. We did a large-scale statistical analysis on short words. We found, for example, even at 70% identity, sequences still have statistically significant number of common pentapeptides. Current CD-HIT is based on this short word statistics. But the short word filters are still limited to certain thresholds. The reasonable limits of clustering thresholds for pentapeptide, tetrapeptide, tripeptide and dipeptide are approximately 70%, 60%, 50% and 40%, respectively.
There is another problem introduced by the greedy incremental clustering. Let say, there are two clusters: cluster #1 has A, X and Y where A is the representative, and cluster #2 has B and Z where B is the representative. The problem is that even if Y is more similar to B than to A, it can still in cluster #1, simple because Y first hit A during clustering process. While this problem could be reduced by multiple-step clustering (see following sections).
CD-HIT-2D comparing algorithm
The above short word filtering and index table can also be used in other sequence comparison tasks, for example, comparing two data sets and reporting the matches between 2 datasets over a certain similarity threshold. This is a very common job, so I developed another program cd-hit-2d for fast comparison of two dataset.
DNA / RNA clustering & comparing
The original CD-HIT was developed for protein clustering. But the short word filtering and index table implementation can also be applied to DNA / RNA. Therefore, I wrote another two new programs cd-hit-est and cd-hit-est-2d. I believe they can be very useful in handling EST sequences.
PSI-CD-HIT clustering
The lowest threshold of CD-HIT is around 40%, in many applications, people need a much lower threshold, like 25%. I am planning develop such application (may be called CD-HIT-LOW, I don’t know yet), but for now, I use PSI-CD-HIT for this purpose.
PSI-CD-HIT is actually a Perl script I wrote, which runs similar algorithm like CD-HIT but using BLAST to calculate similarities. Below are the procedures of PSI-CD-HIT:
- Sort sequences by decreasing length
- First one is the first representative
- Using 1st one blast all remaining sequences, pick up its neighbors that meet the clustering threshold
- Repeat until done
User’s Guide
Installation
Most CD-HIT programs were written in C++. Installing CD-HIT package is very simple:
1. download current CD-HIT at http://bioinformatics.org/cd-hit/, for example cd-hit-2006-0215.tar.gz
2. unpack the file with “tar xvf cd-hit-2006-0215.tar.gz --gunzip”
3. change dir by “cd cd-hit-2006”
4. compile the programs by “make”
5. you will have all cd-hit programs compiled
There are some macros defined in a cd-hi.h that control some basic parameters. I believe, in 99% of the case, that these setting are fine. But you can change them also. I list some of them here:
#define MAX_SEQ 65536
Max length of sequences.
#define MAX_DIAG 133000
This number should be the double of MAX_SEQ.
#define MAX_GAP 65536
Max allowed gap length in dynamic programming subroutine.
#define MAX_LINE_SIZE 300000
Max allowed length of a single line from input FASTA file.
#define MAX_FILE_NAME 1280
Max allowed length of filename.
#define MAX_SEG 50
For large database, the program divides it into several parts,
this number is max allowed No. of parts.
CD-HIT
CD-HIT clusters proteins into clusters that meet a user-defined similarity threshold, usually a sequence identity. Each cluster has one representative sequence. The input is a protein dataset in fasta format and the output are two files: a fasta file of representative sequences and a text file of list of clusters.
Basic command:
cd-hit -i nr -o nr100 -c 1.00 -n 5 -M 2000
cd-hit -i db -o db90 -c 0.9 -n 5, where
db is the filename of input,
db90 is output,
0.9, means 90% identity, is the clustering threshold
5 is the size of word
Choose of word size:
-n 5 for thresholds 0.7 ~ 1.0
-n 4 for thresholds 0.6 ~ 0.7
-n 3 for thresholds 0.5 ~ 0.6
-n 2 for thresholds 0.4 ~ 0.5
Complete options:
-i input input filename in fasta format, required
-o output filename, required
-c sequence identity threshold, default 0.9
this is the default cd-hit's "global sequence identity"
calculated as:
number of identical amino acids in alignment
divided by the full length of the shorter sequence
-G use global sequence identity, default 1
if set to 0, then use local sequence identity, calculated as :
number of identical amino acids in alignment
divided by the length of the alignment
NOTE!!! don't use -G 0 unless you use alignment coverage controls
see options -aL, -AL, -aS, -AS
-b band_width of alignment, default 20
-M max available memory (Mbyte), default 400
-n word_length, default 5, see user's guide for choosing it
-l length of throw_away_sequences, default 10
-t tolerance for redundance, default 2
-d length of description in .clstr file, default 20
if set to 0, it takes the fasta defline and stops at first space
-s length difference cutoff, default 0.0
if set to 0.9, the shorter sequences need to be
at least 90% length of the representative of the cluster
-S length difference cutoff in amino acid, default 999999
if set to 60, the length difference between the shorter sequences
and the representative of the cluster can not be bigger than 60
-aL alignment coverage for the longer sequence, default 0.0
if set to 0.9, the alignment must covers 90% of the sequence
-AL alignment coverage control for the longer sequence, default 99999999
if set to 60, and the length of the sequence is 400,
then the alignment must be >= 340 (400-60) residues
-aS alignment coverage for the shorter sequence, default 0.0
if set to 0.9, the alignment must covers 90% of the sequence
-AS alignment coverage control for the shorter sequence, default 99999999
if set to 60, and the length of the sequence is 400,
then the alignment must be >= 340 (400-60) residues
-B 1 or 0, default 0, by default, sequences are stored in RAM
if set to 1, sequence are stored on hard drive
it is recommended to use -B 1 for huge databases
-p 1 or 0, default 0
if set to 1, print alignment overlap in .clstr file
Output:
The output .clstr file looks like
>Cluster 0
0 2799aa, >PF04998.6|RPOC2_CHLRE/275-3073... *
>Cluster 1
0 2214aa, >PF06317.1|Q6Y625_9VIRU/1-2214... at 80%
1 2215aa, >PF06317.1|O09705_9VIRU/1-2215... at 84%
2 2217aa, >PF06317.1|Q6Y630_9VIRU/1-2217... *
3 2216aa, >PF06317.1|Q6GWS6_9VIRU/1-2216... at 84%
4 527aa, >PF06317.1|Q67E14_9VIRU/6-532... at 63%
>Cluster 2
0 2202aa, >PF06317.1|Q6UY61_9VIRU/8-2209... at 60%
1 2208aa, >PF06317.1|Q6IVU4_JUNIN/1-2208... *
2 2207aa, >PF06317.1|Q6IVU0_MACHU/1-2207... at 73%
3 2208aa, >PF06317.1|RRPO_TACV/1-2208... at 69%
Where,
a “>” starts a new cluster
a “*” at the end means that this sequence is the representative of this cluster
a “%” is the identity between this sequence and the representative
MCD-HIT
MCD-HIT is modified version of CD-HIT. It cuts long proteins into ~400aa pieces to improve short-word filtering efficiency. It is more suitable for datasets that contain proteins of very different lengths and at low clustering threshold <60%.
CD-HIT-2D
CD-HIT-2D compares 2 protein datasets (db1, db2). It identifies the sequences in db2 that are similar to db1 at a certain threshold. The input are two protein datasets (db1, db2) in fasta format and the output are two files: a fasta file of proteins in db2 that are not similar to db1 and a text file that lists similar sequences between db1 & db2.
Basic command:
cd-hit-2d -i db1 -i2 db2 -o db2novel -c 0.9 -n 5, where
db1 & db2 are inputs,
db2novel is output,
0.9, means 90% identity, is the comparing threshold
5 is the size of word
Please note that by default, I only list matches where sequences in db2 are not longer than sequences in db1. You may use options -S2 or -s2 to overwrite this default. You can also run command:
cd-hit-2d -i db2 -i2 db1 -o db1novel -c 0.9 -n 5
Choose of word size (same as cd-hit):
-n 5 for thresholds 0.7 ~ 1.0
-n 4 for thresholds 0.6 ~ 0.7
-n 3 for thresholds 0.5 ~ 0.6
-n 2 for thresholds 0.4 ~ 0.5
More options:
Options, -b, -M, -l, -d, -t, -s, -S, -B, -p, -aL, -AL, -aS, -AS are same to CD-HIT, here are few more cd-hit-2d specific options:
-i2 input filename for db2 in fasta format, required
-s2 length difference cutoff for db1, default 1.0
by default, seqs in db1 >= seqs in db2 in a same cluster
if set to 0.9, seqs in db1 may just >= 90% seqs in db2
-S2 length difference cutoff, default 0
by default, seqs in db1 >= seqs in db2 in a same cluster
if set to 60, seqs in db2 may 60aa longer than seqs in db1
CD-HIT-EST
CD-HIT-EST clusters a nucleotide dataset into clusters that meet a user-defined similarity threshold, usually a sequence identity. The input is a DNA/RNA dataset in fasta format and the output are two files: a fasta file of representative sequences and a text file of list of clusters.
Since eukaryotic genes usually have long introns, which cause long gaps, it is difficult to make full-length alignments for these genes. So, CD-HIT-EST is good for non-intron containing sequences like EST.
Basic command:
cd-hit-est -i est_human -o est_human95 -c 0.95 -n 8
Choose of word size:
-n 8,9,10 for thresholds 0.90 ~ 1.0
-n 7 for thresholds 0.88 ~ 0.9
-n 6 for thresholds 0.85 ~ 0.88
-n 5 for thresholds 0.80 ~ 0.85
-n 4 for thresholds 0.75 ~ 0.8
More options:
Options, -b, -M, -l, -d, -t, -s, -S, -B, -p, -aL, -AL, -aS, -AS are same to CD-HIT, here are few more cd-hit-est specific options:
-r 1 or 0, default 0, if set to 1, comparing both strand (++, +-)
CD-HIT-EST-2D
CD-HIT-EST-2D compares 2 nucleotide datasets (db1, db2). It identifies the sequences in db2 that are similar to db1 at a certain threshold. The input are two DNA/RNA datasets (db1, db2) in fasta format and the output are two files: a fasta file of sequences in db2 that are not similar to db1 and a text file that lists similar sequences between db1 & db2.
For same reason as CD-HIT-EST, CD-HIT-EST-2D is good for non-intron containing sequences like EST.
Basic command:
cd-hit-est-2d -i mrna_human -i2 est_human -o est_human_novel -c 0.95 -n 8
Choose of word size (same as CD-HIT-EST):
-n 8,9,10 for thresholds 0.90 ~ 1.0
-n 7 for thresholds 0.88 ~ 0.9
-n 6 for thresholds 0.85 ~ 0.88
-n 5 for thresholds 0.80 ~ 0.85
-n 4 for thresholds 0.75 ~ 0.8
More options:
Options, -b, -M, -l, -d, -t, -s, -S, -s2, -S2, -B, -p, -aL, -AL, -aS, -AS are same to CD-HIT-2d, here are few more cd-hit-est-2d specific options:
-r 1 or 0, default 0, if set to 1, comparing both strand (++, +-)
CD-HIT-PARA
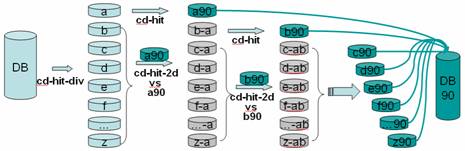
CD-HIT-PARA is a script that runs cd-hit, cd-hit-est in a parallel mode. It splits the input database; runs cd-hit or cd-hit-est in parallel on a computer cluster; and finally merges the outputs into a single file. You can run it as you run cd-hit or cd-hit-est. The input is a protein or DNA/RAN dataset in fasta format and the output are two files: a fasta file of representative sequences and a text file of list of clusters.
Implementation (see figure below)
1. divide input db into many small dbs in decreasing length
2. clusters the 1st db by cd-hit
3. run cd-hit-2d for other dbs against 1st db
4. repeat cd-hit and cd-hit-2d runs till done
5. Combine the results
Basic command:
cd-hit-para.pl -i nr90 -o nr60 -c 0.6 -n 4 -B hosts -S 64, where
-B hosts is a file with available hostnames
-S 64 is the number to split input db into, this number should be several times the number of hosts
PSI-CD-HIT clustering
PSI-CD-HIT clusters proteins into clusters that meet a user-defined similarity threshold, which can be identity or expect value. Each cluster has one representative sequence. The input is a protein dataset in fasta format and the output are two files: a fasta file of representative sequences and a text file of list of clusters
Basic command:
psi-cd-hit.pl -i nr60 -o nr30 -c 0.3
psi-cd-hit.pl -i nr60 -o nr30 -c 0.3 -b hosts
More options:
Options, -l, -d, -s, -S are same to CD-HIT, here are few more psi-cd-hit specific options:
-ce clustering threshold (blast expect), default -1, by default it doesn't use expect threshold, but with positive value, the program cluster sequences if similarities meet either identity threshold or expect value threshold
-L coverage of shorter sequence (aligned / full), default 0
-M coverage of longer sequence (aligned / full), default 0
-R (1/0) use psi-blast profile? default 0, perform psi-blast / pdb-blast type search
-G (1/0) use global identity? default 1, sequence identity calculated as total identical residues of local alignments / length of shorter sequence
-be blast expect cutoff, default 0.000001
-b filename of list of hosts, to run this program in parallel with ssh calls
Incremental clustering
It is easy to make incremental update with cd-hit /cd-hit-2d. For example:
nr is the nr database of last month
month is the new sequences of nr of this month
In last month, you ran:
cd-hit -i nr -o nr90 -c 0.9 -n 5
This month, you can run incremental clustering
cd-hit-2d -i nr90 -i2 month -o month-new -c 0.9 -n 5
cd-hit -i month-new -o month90 -c 0.9 -n 5
cat month90 >> nr90
clstr_merge.pl nr90.clstr month-new.clstr > temp.clstr
cat temp.clstr month90.clstr > this_month_nr90.clstr
This approach is much faster than runing from scratch. It also preserves stable cluster structure.
Hierarchically clustering
With multiple-step, iterated runs of CD-HIT, you perform a clustering in a neighbor-joining method, which generates a hierarchical structure.
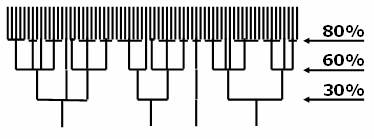
Commands:
cd-hit -i nr -o nr80 -c 0.8 -n 5
cd-hit -i nr80 -o nr60 -c 0.6 -n 4
psi-cd-hit.pl -i nr60 -o nr30 -c 0.3
This way is faster than one-step run from nr directly to nr30. It can also helps correct errors by one-step clustering (see last paragraph in algorithm limitation section).
CD-HIT tools
plot_len.pl
This is a script to print out distributions of clusters & sequences.
Commands:
plot_len.pl input.clstr \
1,2-4,5-9,10-19,20-49,50-99,100-299,500-99999 \
10-59,60-149,150-499,500-1999,2000-999999
where
2nd line are sizes of cluster
3rd line are lengths of sequences
It will print distribution of clusters and sequences :
Size # seq #clstr 10-59 60-149 150-499 500-1999 2000-up
1 266312 266312 36066 103737 103285 22727 497
2-4 208667 81131 1229 14680 44607 20006 609
5-9 156558 24198 118 2148 12026 9388 518
10-19 155387 11681 30 596 5024 5462 569
20-49 176815 6007 6 139 2212 3135 515
50-99 106955 1568 0 24 410 955 179
100-499 154209 896 0 3 124 597 172
500-up 43193 40 0 0 1 14 25
Total 1268096 391833 37449 121327 167689 62284 3084
clstr_sort_by.pl
This script sort clusters in .clstr file by length, size
Commands:
Clstr_sort_by.pl input.clstr no > input_sort.clstr
Where, no means by size of the cluster
clstr_sort_prot_by.pl
This script sort sequences within clusters in .clstr file by length, name, etc.
Commands:
Clstr_sort_prot_by.pl input.clstr id > input_sort.clstr
Where, no means by id of sequences
clstr_merge.pl
It merges two or more .clstr files
Commands:
cd-hit-2d -i db1 -i2 db2 -o db2new -c 0.9 -n 5
cd-hit-2d -i db1 -i2 db3 -o db3new -c 0.9 -n 5
clstr_merge.pl db2new.clstr db3new.clstr > db23new.clstr
clstr_ renumber.pl
It renumbers clusters and sequences within clusters in .clstr file after merge or other operations
Commands:
Clstr_renumber.pl input.clstr > input_ren.clstr
clstr_rev.pl
It combines a .clstr file with its parent .clstr file
Commands:
cd-hit -i nr -o nr90 -c 0.9 -n 5
cd-hit -i nr90 -o nr60 -c 0.6 -n 4
clstr_rev.pl nr90.clstr nr60.clstr > nr60_from90.clstr
psi-cd-hit -i nr60 -o nr30 -c 0.3
clstr_rev.pl nr60_from90.clstr nr30.clstr > nr30_from90.clstr
FAQ
To be added later in a separate document.
References
If you find cd-hit helpful to your research and study, please kindly cite these two references.
- Clustering of highly homologous sequences to reduce the size of large protein databases. Weizhong Li, Lukasz Jaroszewski & Adam Godzik. Bioinformatics (2001) 17:282-283, PDF, Pubmed
- Tolerating some redundancy significantly speeds up clustering of large protein databases. Weizhong Li, Lukasz Jaroszewski & Adam Godzik. Bioinformatics (2002) 18: 77-82, PDF, Pubmed
- Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Weizhong Li & Adam Godzik. . Bioinformatics (2006) 22:1658-1659 PDF, Pubmed