How to get the most out of molecular structure tutorials in Jmol.
available from the
Top Five 3D Molecular Visualization Technologies
at MolviZ.Org


|
Jmol Tutorial Help
How to get the most out of molecular structure tutorials in Jmol. |
available from the Top Five 3D Molecular Visualization Technologies at MolviZ.Org |
|
Please see this separate document.
Select Color
, then Background
, then click on a color.
This will change the background color only until you
Use Select
to select the portion of the molecule whose colors
you wish to change. The number of atoms selected is shown in parentheses,
on the menu, after the word Select
.
After making a selection, use Color
on the menu to apply a plain
color, or a color scheme. These colors will remain only until you
Toggle Spinningbelow the molecule.
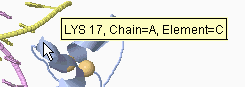 Simply touch (don't click) on an atom with the mouse. Hold the mouse still
over the atom without moving it, which is called "hovering". After a short delay,
a yellow message will appear near the mouse pointer
(see snapshot at right) with an atom identification report.
Clicking on an atom suppresses the hover report -- to see the report,
simply move the mouse away and then back over the atom of interest.
Simply touch (don't click) on an atom with the mouse. Hold the mouse still
over the atom without moving it, which is called "hovering". After a short delay,
a yellow message will appear near the mouse pointer
(see snapshot at right) with an atom identification report.
Clicking on an atom suppresses the hover report -- to see the report,
simply move the mouse away and then back over the atom of interest.
The sequence number in the hover report (17
in the above
snapshot) is that assigned by the
authors of the molecular model
(PDB file)
you are viewing.
It may or may not agree with the numbering in a different model of the same
molecule, or with other published reports.
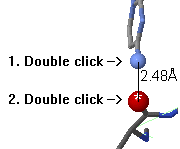 To measure the distance between two atoms, double click on the
first atom. Now, as you touch other atoms, the distances will be
shown on a red line connecting the atoms.
Double click on a second atom to fix the distance report.
To measure the distance between two atoms, double click on the
first atom. Now, as you touch other atoms, the distances will be
shown on a red line connecting the atoms.
Double click on a second atom to fix the distance report.
If you wish to cancel without fixing a distance report, click where there is no atom (on the background within JSmol). If you wish to remove one already fixed report, double click on each of those two atoms again. If you wish to remove all distance reports, click the View button to refresh the molecular view.

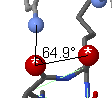 You may also measure angles (or dihedral angles) by double-clicking
on the first atom, single clicking on one (or two) subsequent atoms, and double clicking
on the final atom.
You may also measure angles (or dihedral angles) by double-clicking
on the first atom, single clicking on one (or two) subsequent atoms, and double clicking
on the final atom.
If you wish to explore the molecules in this tutorial on your own,
you can do so with the links that display them in FirstGlance in Jmol,
the easiest tool for general exploration of 3D macromolecular structures.
These links are in the How To
section at the bottom of each
control panel in the tutorial, after you click SHOW LIST OF MOLECULES.
You may also go directly to firstglance.jmol.org to initiate exploration of any available molecule of your choosing.
The Jmol Menu provides access to a wide range of Jmol's capabilities. The menu is good for exploring what Jmol can do, or for making a small number of changes to the molecular view. However, it is too cumbersome for extensive, self-guided exploration, which can be done much more easily in FirstGlance in Jmol (previous section, above).
Jmol's Console is where you can enter typed commands to change the molecular view while using a tutorial. For those who have learned some of Jmol's command language, this is more efficient than using Jmol's menu. The Console can be opened from Jmol's menu.