|
What's Inside Outside Links |
|
Main»Home Page
MFold Interface Perl/Tk GUI Interface for Dr. Michael Zuker's MFold. Allows multiple sequence submission/result retrieval. Can obtain sequences by entering GI number or directly entering sequence data. All non-sequence characters will be removed from box (only ACGTU or acgtu will be accepted, allows for pasting of numerical data). Will only work with 'immediate' jobs, so sequence length is limited to 800 nucleotides. If selected start and end are over 800nt, end will be automatically adjusted to 800nts past the start site. If an end site is selected past the actual sequence end, the end of the sequence will be automatically selected. Directory selection will allow for a main directory for output data. Program will create subdirectories based on given sequence name, start, and end nucleotides (Ex. Name = CVB3-GA, Start = 1, End = 742: CVB3-GA_1-742) Sequence names should only contain letters, numbers, hyphen, and underscore. Program currently provides limited error checking, more will be enabled later. After selecting sequences for folding, program will sequentially submit data to MFold and wait for processing. Once finished, it will download selected result files (currently limited to CT, Vienna, JPG, SS Count, P Num, and H Num files) to subdirectory and then procede to the next available sequence. The open command prompt will show sequence status for the current run (current sequence/step of download process). Some files take longer by the server to process (JPG Zip File). If file is not available on first try, the program will pause for 20 seconds (status displayed in command prompt) and then make up to four more attempts to download the file. Any recommended changes, as well as bugs, are welcome at schu1321@bioinformatics.org For additional development, the author can be contacted as can the Bioinformatics Group at the University of Minnesota. This is a student group at the University of Minnesota that develops open-source software for staff and faculty, as well as other interested parties as time permits. Screenshots: 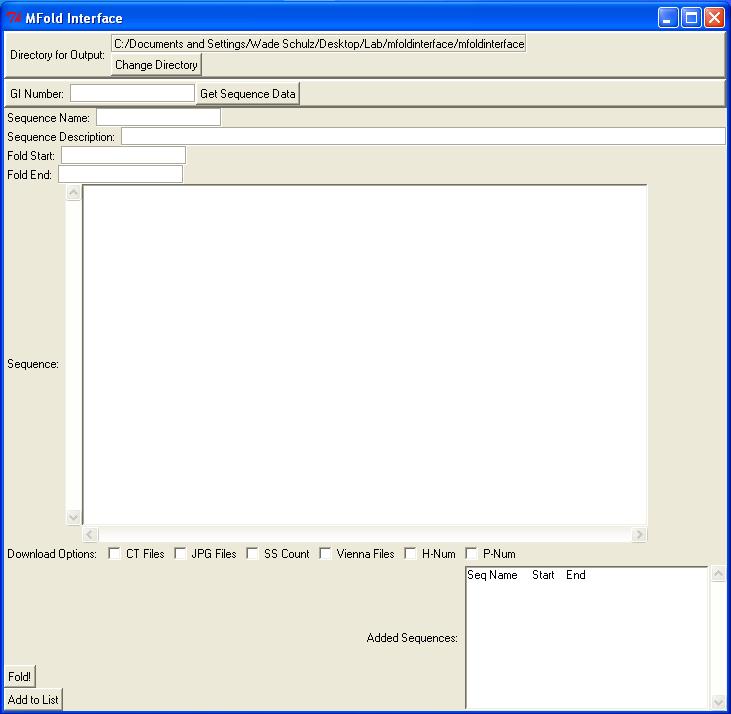 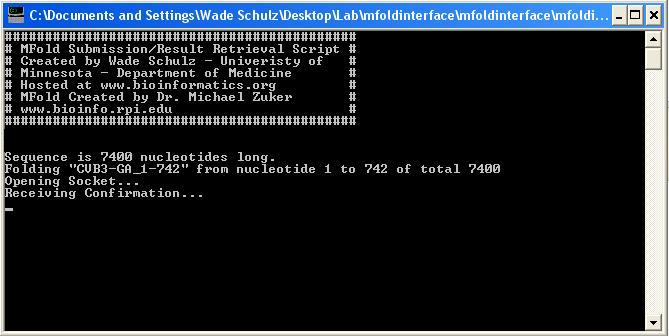 |