Primary information of p53 gene
1.INTRODUCTION
2.HISTORY
p53 was identified in 1979 by Arnold Levine,David Lane and William Old,working at Princeton University, Dundee University (UK) and Sloan-Kettering Memorial Hospital, respectively. It had been hypothesized to exist before as the target of the SV40 virus, a strain that induced development of tumors.Although it was initially presumed to be an oncogene, its character as a tumor suppressor gene was revealed in 1989.In 1993, p53 protein has been voted molecule of the year by the Science magazine
3. GENE
4. STRUCTURE
The p53 protein is a phosphoprotein made of 393 amino acids. It consists of four units (or domains):
A domain that activates transcription factors.
A domain that recognizes specific DNA sequences (core domain).
A domain that is responsible for the tetramerization of the protein.
A domain that recognized damaged DNA, such as misaligned base pairs or single-stranded DNA.
Wild-type p53 is a labile protein, comprising folded and unstructured
regions
which function in a synergistic
manner (Bell et al. 2002).p53 protein
has been
voted molecule of the year.
5. MECHANISM
It plays an important role in cell cycle control and apoptosis. Defective p53 could allow abnormal cells to proliferate, resulting in cancer. As many as 50% of all human tumors contain p53 mutants.
In normal cells, the p53 protein level is low. DNA damage and other stress signals may trigger the increase of p53 proteins, which have three major functions: growth arrest, DNA repair and apoptosis (cell death). The growth arrest stops the progression of cell cycle, preventing replication of damaged DNA. During the growth arrest, p53 may activate the transcription of proteins involved in DNA repair. Apoptosis is the "last resort" to avoid proliferation of cells containing abnormal DNA.
The cellular concentration of p53 must be tightly regulated. While it can suppress tumors, high level of p53 may accelerate the aging process by excessive apoptosis. The major regulator of p53 is Mdm2, which can trigger the degradation of p53 by the ubiquitin system.
Target Genes
p53 is a transcriptional activator, regulating the expression of Mdm2 (for its own regulation) and the genes involved in growth arrest, DNA repair and apoptosis. Some important examples are listed below.
Regulation of p53
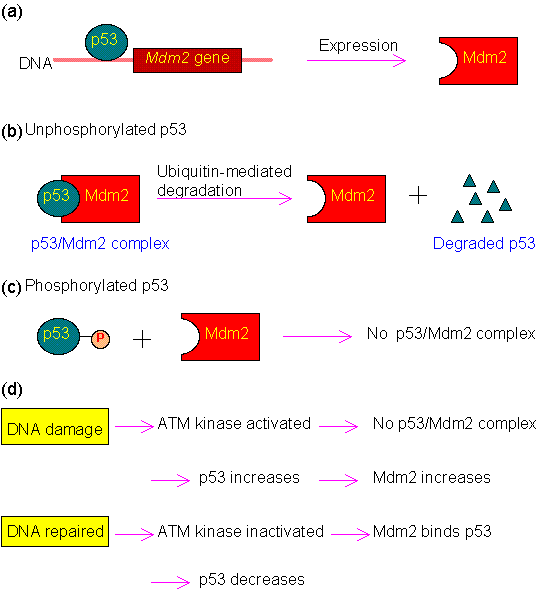
As mentioned above, p53 is mainly regulated by Mdm2. The regulation mechanism is illustrated in the following figure.
Figure 1.0. Regulation of p53.
(a) Expression of Mdm2 is activated by p53.
(b) Binding of p53 by Mdm2 can trigger the degradation of p53 via the ubiquitin system.
(c) Phosphorylation of p53 at Ser15, Thr18 or Ser20 will disrupt its binding with Mdm2. In normal cells, these three residues are not phosphorylated, and p53 is maintained at low level by Mdm2.
Roles of p53
The roles of p53 in growth arrest and apoptosis are illustrated in Figure 4-H-6. p53 is also directly involved in DNA repair. One of its transcriptional target gene, p53R2, encodes ribonucleotide reductase, which is important for both DNA replication and repair. p53 also interacts directly with AP endonuclease and DNA polymerase which are involved in base excision repair.
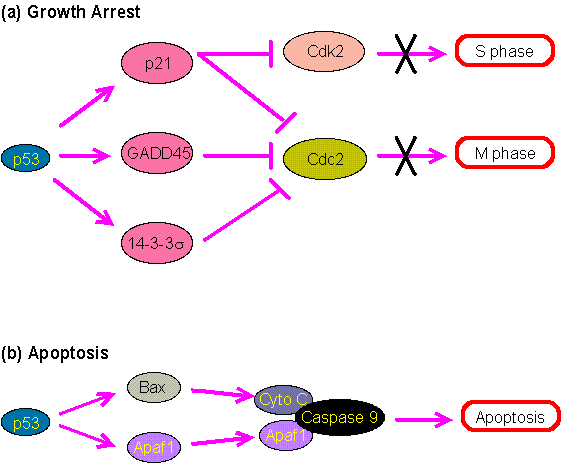
Figure 2.0. The roles of p53 in growth arrest and apoptosis.
(a) The cell cycle progression into the S phase requires the enzyme Cdk2, which can be inhibited by p21. The progression into the M phase requires Cdc2 which can be inhibited by p21, GADD45 or 14-3-3s. p53 regulates the expression of these inhibitory proteins to induce growth arrest.
(b) Apoptosis can be induced by the binding of Caspase 9 to cytochrome c and Apaf1. p53 may activate the expression of Apaf1 and Bax. The latter can then stimulate the release of cytochrome c from mitochondria (see Mitochondria, Apoptosis and Aging).
6. ROLE IN DISEASE
If the p53 gene is damaged, tumor suppression is severely reduced. People who inherit only one functional copy of p53 will most likely develop tumors in early adulthood, a disease known as Li-Fraumeni syndrome. p53 can also be damaged in cells by mutagens (chemicals, radiation or viruses), increasing the likelihood that the cell will begin uncontrolled division. More than 50 percent of human tumors contain a mutation or deletion of the p53 gene.
In health p53 is continually produced and degraded in the cell. The degradation of p53 is, as mentioned, associated with MDM-2 binding. In a negative feedback loop MDM-2 is itself induced by p53. However mutant p53s often don't induce MDM-2, and are thus able to accumulate at very high concentrations. Worse, mutant p53 protein itself can inhibit normal p53 (Blagosklonny, 2002).
7. POTENTIAL THERAPEUTIC USE
In-vitro introduction of p53 in to p53-deficient cells has been shown to cause rapid death of cancer cells or prevention of further division. It is more these acute effects which hopes rest upon therapeutically (McCormick F, 2001). The rationale for developing therapeutics targeting p53 is that "the most effective way of destroying a network is to attack its most connected nodes". P53 is extremely well connected (in network terminology it is a hub) and knocking it out cripples the normal functioning of the cell. This can be seen as 50% of cancers have missense point mutations in the p53 gene, these mutations impair its anti-cancer gene inducing effects. Restoring its function would be a major step in curing many cancers (Vogelstein et al 2000).
Various strategies have been proposed to restore p53 function in cancer cells (Blagosklonny,2002).A number of groups have found molecules which appear to restore proper tumour suppressor activity of p53 in vitro. These work by altering the conformation of mutant conformation of p53 back to an active form. So far, no molecules have shown to induce biological responses, but some may be lead compounds for more biologically active agents. A promising target for anti-cancer drugs is the molecular chaperone Hsp90, which interacts with p53 in vivo.
Adenoviruses rely on their host cells to replicate, they do this by secreting proteins which compel the host to replicate the viral DNA. Adenoviruses have been implicated in cancer-causing diseases, but in a twist it is now modified viruses which are being used in cancer therapy. ONYX-015 (dl1520, CI-1042) is a modified adenovirus which selectively replicates in p53-deficient cancer cells but not normal cells (Bischoff, 1996). It is modified from a virus that expresses the early region protein, E1B, which binds to and inactivates p53. P53 suppression is necessary for the virus to replicate. In the modified version of the virus E1B has been deleted. It was hoped that the viruses would select tumour cells, replicate and spread to other surrounding malignant tissue thus increasing distribution and efficacy. The cells which the adenovirus replicates in are lysed and so the tumour dies.
Preclinical trials using the ONYX-015 virus on mice were promising however clinical trials have been less so. No objective responses have been seen except when the virus was used in combination with chemotherapy (McCormick, 2001). This may be due to the discovery that E1B has been found to have other functions vital to the virus. Additionally its specificity has been undermined by findings showing that the virus is able replicate in some cells with wild-type p53. The failure of the virus to produce clinical benefits may in large part be due to extensive fibrotic tissue hindering virus distribution around the tumour (McCormick, 2001).
8. REFERENCES
Bell S, Klein C, Muller L, Hansen S, Buchner J. (2002). p53 contains large unstructured regions in its native state. J Mol Biol, 322:917-927
Bischoff JR, Kirn DH, Williams A, Heise C, Horn S, Muna M, Ng L, Nye JA, Sampson-Johannes A, Fattaey A, McCormick F. (1996). An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science, 274:373-376
Blagosklonny, MV. (2002). P53: An ubiquitous target of anticancer drugs. International Journal of Cancer, 98:161-166
McCormick F. (2001). Cancer gene therapy: fringe or cutting edge? Nat Rev Cancer, 1:130-141
Strachan T, Read AP. (1999). Human Molecular Genetics 2. Ch. 18, Cancer Genetics
Vogelstein B, Lane D, Levine AJ. (2000). Surfing the p53 network. Nature, 408:307-310