Modifying annotations
To edit a * residue annotation the menu
item  of the
context menu must be clicked. Alternatively the residue underlining in the alignment panel can be double clicked.
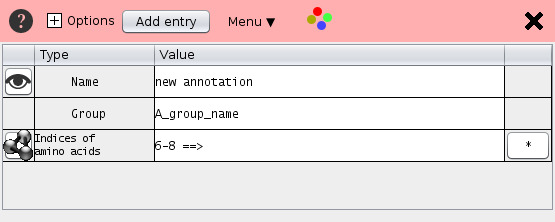
The annotation data is shown as a table. The rows contain the name, the group name, the residue positions and additional annotation.
of the
context menu must be clicked. Alternatively the residue underlining in the alignment panel can be double clicked.
The annotation data is shown as a table. The rows contain the name, the group name, the residue positions and additional annotation.
-
The most important entry is Positions which defines the the selected sequence positions.
Here are some examples of valid entries
-
The expression "1,3,4,6,101-103,110-112"
selects the residue indices 1,3,4,6,101,102,103,110,111,112.
Instead of the commas, spaces can by used.
-
The expression
"+2 1 3 4"
selects the residues 3,5, and 6 because the preceding +2 adds an offset of 2 to all positions. The "+"-sign is the first character in this expression.
For the user interface the first residue has the index "1" whereas internally STRAP starts counting at zero.
A negative offset of "2" is achieved by a leading "+-2".
-
The expression
"1:G-3:G 5:G"
selects the residues with the pdb-numbers 1,2, 3 and 5 of the chain G.
If the protein has only one chain the chain identifier can be omitted like
"1:-3: 5:"
-
Referring to nucleotide positions:
When the toggle button is pressed (default state) the indices refer to amino acid positions.
Otherwise the positions indicate nucleotides in case the protein is translated from a nucleotide sequence.
-
To any group an atom specification can be appended.
This allows to select certain atoms before changing their style in the 3D-viewers.
Even though protein 3D viewers usually have a specific language for atom selection,
the atom expression used here is defined by STRAP and works for all 3D-viewers.
for example "10:-20:.CA.CB" narrows the selection to the atoms CA and CB for the residues 10 to 20.
A later command such as "3D_spheres" would affect only c-alpha and c-beta atoms.
There may be more than one "Atoms"-entries. Each "Atoms" specification take effect only on the following 3D-style
commands but not on the previous.
Asterisk can be used as a wild card like "*.CA" or "10:-20:.C".
For the proper atom identifiers have a look at the PDB file.
- Atoms
Certain Atoms can be specified by an expression like ".CB.CA" which means only Cα and Cβ atoms.
The next 3D-command will act on these rather than on all atoms of the amino acid.
When sending the commands to a 3D-program, the rows are processed sequentially.
If there comes yet another Atoms-row,
the previous atom-specification is replaced by the new one.
- Name Each selection has a name. Un-checking the check box deactivates the residue annotation.
- Group Several selections may be bundled in one group e.g. "active site"
 : TeXshade commands. See menu Export in the file menu.
: TeXshade commands. See menu Export in the file menu.- Note, Remark: Free text. URLs are clickable. Supports cross-links like PDB:1ryp, PUBMED:0815. The list of databases can be changed by Ctrl-click the cross-link.
- Balloon: The balloon text appears when the mouse is over the highlighted residues.
Drag and drop: Rows can be reordered with the mouse or can be dropped on other residue selections.