Network reconstruction tutorial
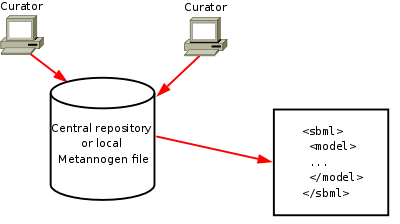
|
 .
The left upper panel contains the networks "KEGG", tab "RECON1" and tab "EHMN" as a browsable tree.
The network components in these tree can be used to define reactions for the network to be reconstructed.
.
The left upper panel contains the networks "KEGG", tab "RECON1" and tab "EHMN" as a browsable tree.
The network components in these tree can be used to define reactions for the network to be reconstructed.
Creating Datasets
Open the KEGG network tree (first tab in the upper left panel) and open the of an enzymatic reaction, i.e. expand a pathway node and chose one of the reactions. Then, click the menu item " New dataset". A new dataset for this enzymatic reaction is created.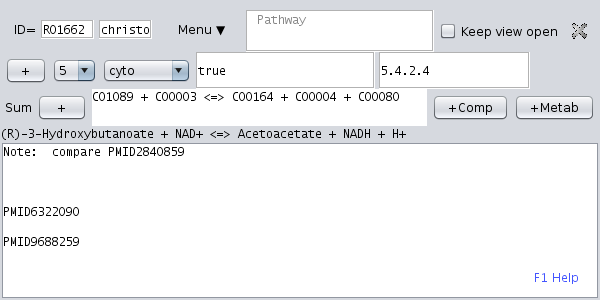 |
Compartments
In the following we are going to explain two different ways to specify compartments for biochemical conversions. For this purpose, create a dataset with an enzymatic reaction. When creating or modifying datasets you can always check, what reaction objects would be generated from the respective dataset. Find the "Menu▼" pull down menu within the dataset form. Use the sub-menu "View" to get a list of all reactions defined by the dataset.Compartment suffices after the metabolite identifiers
Append @cyto (at-sign) and the compartment ID to each metabolite identifier in the equation. Then check the list of reactions. This method is generally applicable. It works even for processes at compartment borders as the following example.Reactions that involve more than one compartment
Create a transport process for NH3 from "cyto" to "endo". you could type the equation:NH3@cyto <=> NH3@endoIf you have loaded the KEGG network, Metannogen will replace NH3 by C00014 as you type. Please learn from this example that transport processes over membranes are just biochemical reactions where the sub-cellular locations of the metabolites are not identical.
Compartments by choice menu
Appending the compartment after each metabolite is tedious and unnecessary if all metabolites have the same compartment. In the following we will present a more convenient method. First create a dataset with a biochemical equation. Do not add compartment suffices. Instead find the compartment choice menu at the top of the dataset form. Select . Now check the created reactions. All metabolites inherit the selected compartment, in this case "cyto". If the respective enzyme would also be active in other compartments such as "mitMx" at the same time, further compartments may be specified. Please generate an additional compartment choice menu with the button and select a compartment . If you look at the list of generated reactions you will find two reactions, one for cyto and one for mitoMx. This method has two advantages over the suffix-method:- Less typing
- Several localized reactions are defined by one single dataset
Unspecific reactions
Motivation: Some reactions or transporters act not on one specific metabolite, but on a variety of different metabolites. When recording conversions of unspecific enzymes or carriers, curators could create several datasets with slightly different biochemical equation. All other information apart from the reaction would be identical. Redundancy, however, should be avoided and typing must be reduced.Solution: To avoid creation of different datasets, can be used. To try brace expansion create a transport dataset with the following reaction string:
{ Alanine Proline}@cyto <=> { Alanine Proline}@mito
Look at the list of created
reaction objects. Add further
metabolites of your choice and
compare the list of created
reactions again.
You see that in this example the first group of metabolites is exactly the same as the second one.
Consequently, you may substitute the second group by "{}". There must not be a space between the two braces:
{ Alanine Proline}@cyto <=> {}@mito
Make up an example where the two groups differ such that the second group cannot be abbreviated by "{}". (Hint: Transaminases, Desaturases of Fatty acids of different length). Try what happens if the number of elements in each group is not identical.
For transport processes the subcellular localization of the metabolites are written as suffices. Here brace expansion can also be applied. The following describes the transmembranal glucose transport across the endoblasmic reticulum membrane and across the cell membrane.
C00031@{cyto cyto} <=> C00031@{ext endo}
It expands to
C00031@cyto <=> C00031@ext
C00031@cyto <=> C00031@endo
An attribute declaration (See Attributes) like $FUNCTION="Metabolism" is inherited by all reactions expanded from the braces. It might be necessary to assign attributes only to one specific reaction. With the example above one might want to create an attribute only for the reaction with Alanine but not with Proline. In this case use the member in the parentheses as a suffix: $FUNCTION{Alanine}="Metabolism"
See Metannogen brace expansion for more.
Reaction Attributes
The multi-line text field contains free text which does not to be structured in a certain way. Usually, it is not possible to include information from this text-field in the exported model file. However, content that is captured in form of a variable will be available for further data processing. Add a simple variable declaration in the multi-line text field. If you know programming languages like or you will be familiar with the syntax.
$THE_FUNCTION=break-down
You can use any variable name starting with a letter and consisting of letters, digits and underscore.
It is recommended to register variable names in Menu-bar>Customize>Variables.
The advantage is that registered variables are color-highlighted.
This allows one to notice when variables are mistyped.
To refer only to the reaction in one compartment add a compartment suffix:
$THE_FUNCTION@cyto=break-down
or several compartments separated by comma
$THE_FUNCTION@cyto,endo=break-down
For text containing white space use quotes:
$THE_FUNCTION@cyto="break down"
Obtain the list of reaction objects of the dataset and observe what reaction[s] the attributes are assigned to.
The variable content can be used for the data export.
See Tutorial SBML-Export.