back to STRAP: multiple sequence alignment
also visit 3D-superposition of proteins and
3D-superposition of proteins and
Dialog protein structure Viewer and
Jmol and
PyMol and Strap
PyMol is a very powerful full featured three-dimensional protein viewer.
It has a comfortable graphical user interface but it can also be controlled by commands.
Therefore it can easily be controlled by STRAP.
STRAP simply sends commands to PyMol via the standard input stream.
When a protein structure is opened STRAP sends the command to PyMol for loading a protein structure file.
Alignment cursor
Picking an atom while the control and shift key is hold down, moves the alignment cursor to the respective residue.
Mouse selection
When residues are marked with the mouse in STRAP an atom selection is created in PyMol.
The user can change the rendering style of the underlined residues.
Named selections
In STRAP a set of residues can be chosen and text commands assigned to this residue selection.
The assigned text is viewed in a table. Consider the following residue selection:

On pressing the "add"-button The following form comes up:

In the left list the user selects PyMol and in the right list a PyMol command as for instance
hide everything, RESIDUES; show spheres,RESIDUES
On pressing the Pymol-label the command is interpreted by Pymol.
Before sending, the variable RESIDUES is replaced by a text expression that denominates the selected residues by STRAP.
The command line can be changed by the user at any time.
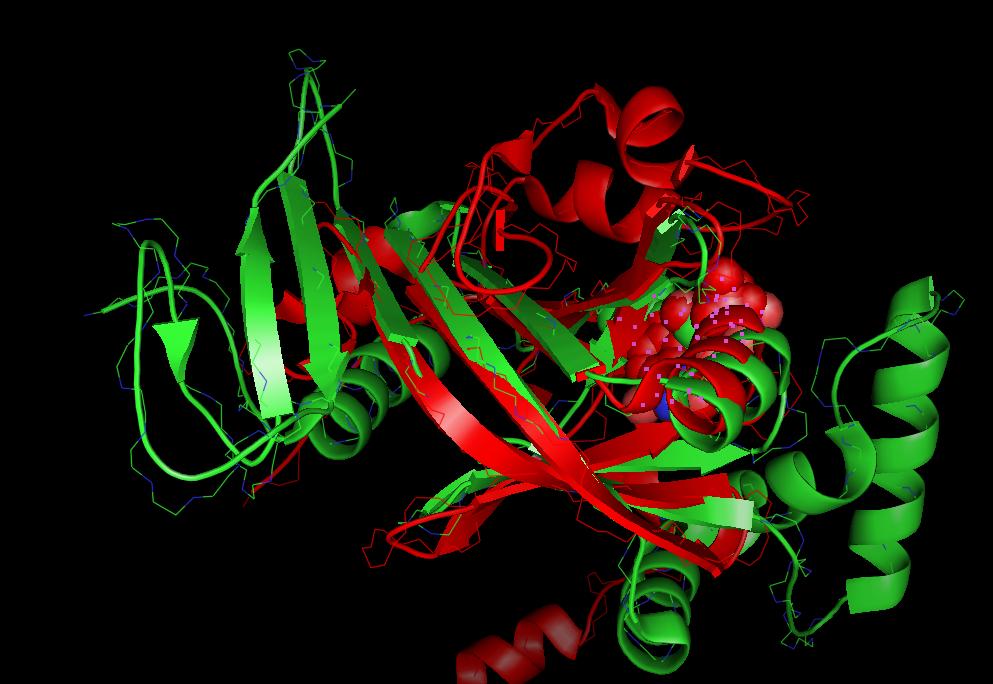
Visualization of protein superpositions
Pymol is currently the only protein viewer which is used in Strap to display 3D- superpositions:
Installation of PyMol
PyMol is obtained from http://pymol.sourceforge.net/ .
The installation is straight forward.
Other Pymol sites
A nice introduction is found at http://www.rubor.de/bioinf/ .
Last modified: Thu Mar 5 21:58:31 Local time zone must be set--see zic manual page 2009