| B | Logo |
Table of content:
How to fill in the form:
Job Name
Sequence Type
Sequence Format
Background frequencies
Choose region
Logo properties
Algorithm:
Type 1 logo
Type 2 logo
Statistical test
=============================================
How to fill in the form:
Input a meaningful name for your job (for example, Ecoli_RBS) will. If you don't input anything, the program will produce a temporary name automatically.
DNA or RNA: like ACGTCGTAGGAAATC or AGCUCCCUUUAGCAAUC;
Protein: like ADGHKLIIASS (using one-letter code);
Translate to AA: create amino acid logos using their coding sequences;
Codon: inputs are coding sequences. The background frequency of nucleotides on the three positions of codon was calculated separately.
multi-fasta, like this:
>seq1
TTGGTTTGGTCAATGCTACAAGAAGATCAGG
AAGTACACGAAGAGAATATGTAATTGGTTTG
>seq2
GTCAATGCTACAAGAAGATCAGGAAGTACAC
GAAGAGAATATGTAATTGGTTTGGTCAATGC
>seq3
TACAAGAAGATCAGGAAGTACACGAAGAGAA
TATGTAATTGGTTTGGTCAATGCTACAAGAA
>seq4
GATCAGGAAGTACACGAAGAGAATATGTAAT
TGGTTCAATGCTACGCTACAAGAAGTGGTTC
...(more)...
one sequence one line, like this:
TTGGTTTGGTCAATGCTACAAGAAGATCAGG
AAGTACACGAAGAGAATATGTAATTGGTTTG
GTCAATGCTACAAGAAGATCAGGAAGTACAC
GAAGAGAATATGTAATTGGTTTGGTCAATGC
TACAAGAAGATCAGGAAGTACACGAAGAGAA
TATGTAATTGGTTTGGTCAATGCTACAAGAA
GATCAGGAAGTACACGAAGAGAATATGTAAT
TGGTTCAATGCTACGCTACAAGAAGTGGTTC
AATGCTACGCTACAAGAAGATCAGGTGGTTC
AATGCTACGCTACAAGAAGATCAGGGTACCC
Calculated from your sequences: the program will calculate the background frequency of every symbol from the sequences you have inputted. When the sequences were codons, the background frequency of nucleotides could be calculated separately in the three positions of codon.
Input yourself: input the background frequency of every symbol by yourself. For DNA or RNA, input the frequencies in the order of A, C, G, T. For proteins, input by this order: G, S, T, Y, C, Q, N, K, R, H, D, E, A, V, L, I, P, W, F, M. For codons, input by this order: A1 C1 G1 T1 A2 C2 G2 T2 A3 C3 G3 T3, where 1, 2, 3 are the three positions of the codon. Separate values with commas. The summary of the values should be 1 for DNA/RNA or proteins and 3 for codon.
Full length of your shortest sequence: If you have inputted sequences with different length, the program will create a logo with number of positions equal to the shortest length of your sequences.
Input yourself: You can choose to create a logo from a specific region on your inputted sequences. The first letter of your sequence is numbered one.
Logo type: The type 1 logo is similar to WebLogo and the type 2 logo was designed to shown sequence bias. The over-represented symbols were stacked above zero and under-represented symbols under zero. The height of every symbol is proportional to its information content.
Middle height (Pixel): set the height of the figure. A high value will make the figure clearer and bigger.
Information Content: set the maximum and minimum values of y axis. For a type 1 logo, Y min should be 0. For a type 2 logo, Y min should be negative and Y max should be positive.
Character width: the width of every symbol. A larger value will make the figure wider.
Image file format: could be jpg, gif or tif.
Statistical test: When a Statistical test is used, a symbol with a P-value larger than a threshold (default is 0.05) will be colored gray. Using a test will make the program slower.
Algorithm
Information content was calculated for each position of sequences using the formula:
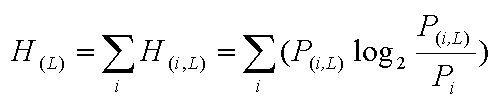
where L is the position in the sequences; i are symbols (A, T, C and G for nucleotides or 20 amino acids for protein sequences); P(i,L) is the average probability of symbol i at position L; and Pi is the background probability of symbol i. H(i,L) is positive when P(i,L) is bigger than Pi, and negative when P(i,L) is smaller than Pi.
For type 1 logo, the total height of the symbols in a position L is equal to H(L), and the height of every symbol i is proportional to the its observed frequency.
For type 2 logo, all the letters from each stack are ordered from the biggest H(i,L) to the smallest, and all letters with a positive H(i,L) are stacked above zero. The height of a symbol i equals to H(i,L).
For every symbol in a position of sequences, chi square test or Fisher's exact test was used to evaluate the statistical significance of the difference between the frequency of that symbol in the position and the background (expected) frequency of that symbol. The program chose chi square test in large samples (number of sequences > 400, observed or expected number of the symbol in each position > 20) and Fisher's exact test in small samples (other conditions).
The difference was assumed to be significant if P-value was less than a threshold (the default value is 0.05 and this can be modified by user).