| CAT-O: conCATenate Orthologous sequences |
| Home | About | Tutorial | Contact | ||
| TUTORIAL | ||
| Home page | ||
It displays a list of parameters required for the search (Fig 1). For finding common orthologous sequences, various Blast parameters are needed, namely type of search (Protein or DNA), filter for low complexity regions and masking, expectation value (by default 10), word size, matrix and gap cost. For concatenation input boxes for positivity (Percentage of similar residues in the alignment), identity (Percentage of identical residues in the alignment) and coverage (Percentage of alignment length to query length) are provided. By default the values are 50 for all. 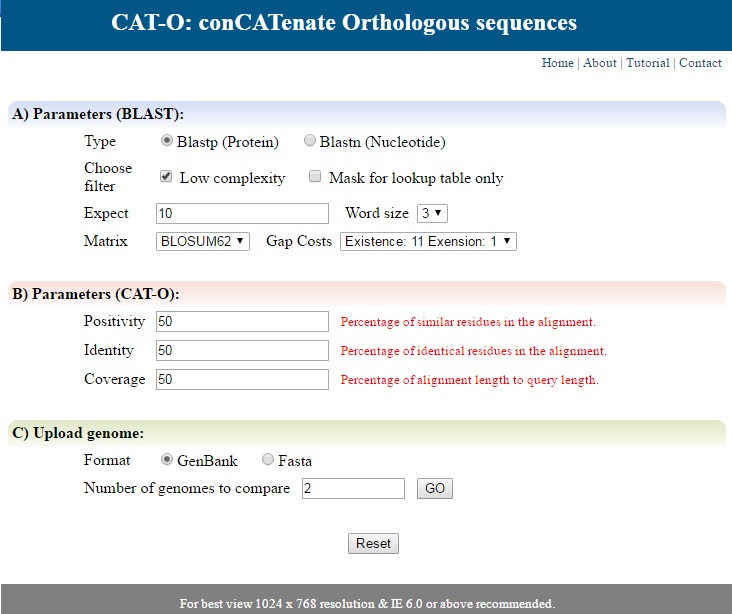 | ||
| Fig 1. Home page | ||
Select the format of file (Genbank or Fasta) and enter the number of genomes to be uploaded. Click Go and browse your files. Click reset button if you want to reset the parameters or submit to submit the form. | ||
| Result page | ||
| Result page consists of two main headings : | ||
1) View Summary table for orthologs - By default this result is displayed (Fig 2). It shows a matrix containing number of common orthologs between all pair of genome files. Clicking on the number will display a table depicting id's of corresponding orthologs. 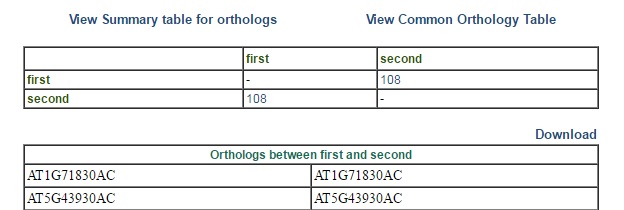 | ||
| Fig 2. Summary page | ||
2) View Common Orthology Table - Here you can view common orthology of a genome with every other genome. You can also go for phylogenetic profile, unique accessions and duplicate accessions of each file by clicking on respective buttons (Fig 3). 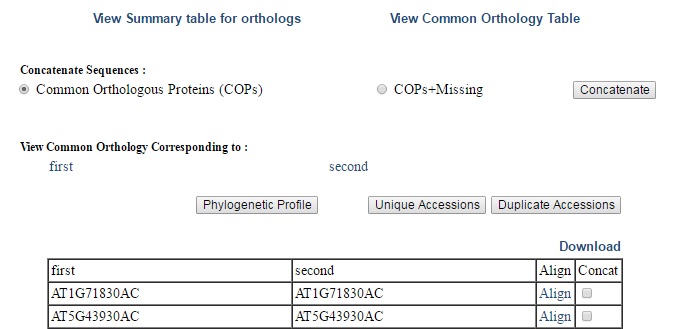 | ||
| Fig 3. Common Orthology page | ||
The Align link next to each row in result table will display the sequence alignment for the corresponding sequence id's (Fig 4).  | ||
| Fig 4. Alignment | ||
For concatenation you can select the check boxes at the end of each row. Under the concatenate sequences heading click on a radio button either for common orthologous proteins (COPs) or COPs+Missing data concatenation. For later you can enter the missing percentage which is 70 by default. On clicking concatenate button will display the required concatenated sequences (Fig 5). In case of DNA, you can also fetch sequences based on positions of triplet codon by selecting radio buttons provided for first-second position, only first position or only second position. 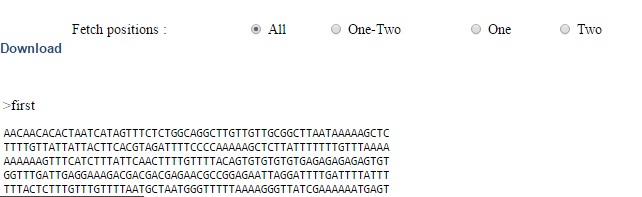 | ||
| Fig 5. concatenation | ||
The results displayed on any page can be downloaded by clicking Download link. | ||
| For best view 1024 x 768 resolution & IE 6.0 or above recommended. |