
A short walk through the program
What you can do with the current beta version of jannotatix, to illustrate why we wrote it
This text describes a sample application of jannotatix, step-by-step.
Start the program
Click on download on the left side and run the program, either with webstart or directly by downloading the .jar-file.
When run for the first time, it will ask for some plugins to be
downloaded from the website. Select Plantcare, Transfac and AlignACE and
wait until the program has finished downloading them. You can add
additional plugins at any time by clicking
Download sample files and open them from your harddisk
We have put two files on this webserver: some sequences in fasta-format and some features for them in GFF-format. Right-click on the two links and save the files somewhere on your harddisk. (Webstart users simply click here to open both the program and the sample files automatically from your browser).
Click
| Screenshot of the sample features |
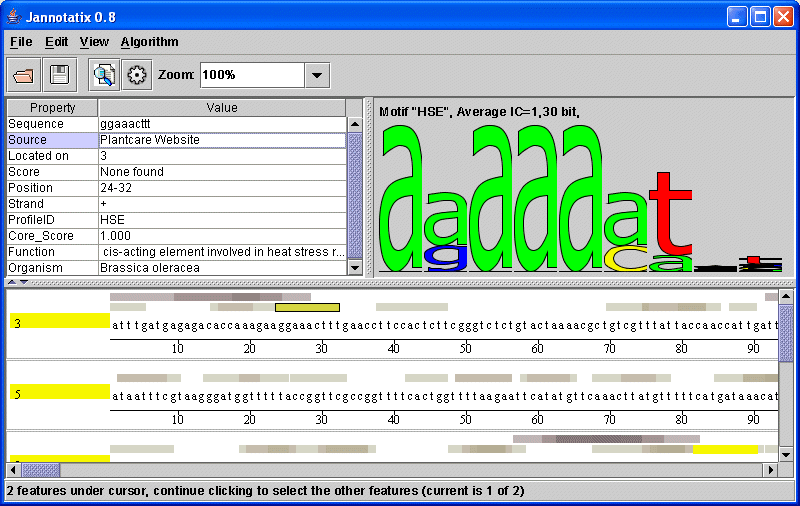
|
Try clicking on a feature. You will see that in the top window, on the left, properties of this feature are shown. They indicate...
-
The conkrete sub-sequence that this feature is covering
-
The source of this feature, i.e. the program or website that generated it
-
The name of the sequence this feature is located on (our sample sequences sometimes only have a simple number as a name)
-
A score, something the source-program attributed to this feature to indicate how well it corresponds to some model.
-
The strand the feature is located on
-
The position, given in numbers, on the sequence
-
And some optional data like the ProfileID (for motif discovery programs, all feature that belong to the same motif should share the same ProfileID. It they have one, a motif logo is calculated and displayed on the right side). If a program generates more than one score, they are also listed, prefixed by ProfileScore_ and then any identifier (AlignACE, for example, that we used for the sample file, generates scores that are called MAP, so the property is named ProfileScore_MAP).
Whenever you click on a feature, all the other, aligned features that share the same ProfileID will be highlighted in yellow. You can use the Zoom-function from the toolbar to see all highlighted features.
If a feature was generated by a motif prediction/discovery program, then you should see a motif logo on the right side. A logo is a visual representation of many aligned features: The higher the letter, the less noise can is found at a position. Therefore, if a logo consists of many small letters, it is not a very good one, and its Information Content (IC) is very low. The maximum value for an Information Content is 2 bits at a given nucleotide position, the score that you can see above the logo is the value averaged over all positions.
Some programs generate a wealth of additional information. PlantCare, in our example, tells you the organism, where this element was found, and some rough classification of its function (see screenshot).
Apply algorithms to generate new features
Click on
| Screenshot of the empty sequences |
|
|
Click on
On the next tab, you can supply a handful of parameters, I suggest
selecting
| The Algorithm-Run Dialog |
|
|
You can run additional algorithms by selecting them from the menu. Try AlignACE, for instance, which is a motif discovery program, that is run on your own computer (we only supply binaries for Windows and Linux), without any HTTP-transfers involved.
Analyse the results
This is something which is not at all completed. You can try View - Motifs but this is very slow (you have to wait very long until the results show up on the screen) and merely lists all motifs across all tracks, sorted by average Information Content. We want to add here a couple features, like direct linking from the motif to the sequence view.
| Screenshot of the motif viewer |
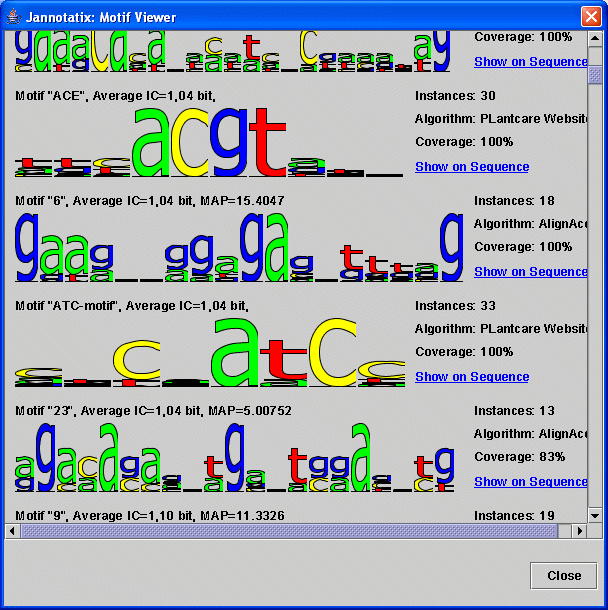
|
Then you will need more tools: filtering very common motifs would be nice, just as comparing the tracks against some kind of reference track, to be able to benchmark an algorithm against others (this is halfway finished in the source code). Unfortunately, Jannotatix is still in early beta stage, these functions will take some weeks or months to complete.
by Maximilian Haeussler