Preparing document for printing:
When printing, backgrounds are automatically turned to white.
Metannogen
Overview
Metannogen is a customizable graphical work-bench to facilitate curation of biological networks for a single user or for a team of data curators. It consists of a graphical browser for metabolic networks and an editor for user defined collections of biochemical equations. Metannogen can be used in two ways:- Reconstruction of metabolic networks
- Annotation of existing metabolic networks given as SBML
1. Annotation of SBML-files
Metannogen allows to load biological models as SBML. These files are not changed by Metannogen, but Metannogen lets the user add information to reactions. The annotation may be free text which does not affect the exported SBML or it can provide information which is included into the exported SBML file.- Free text annotations not influencing the model: The user can add notes, comments including hyper-references, links to pubmed IDs etc.
- Annotations that are included into the output SBML: The output SBML is generated from the read-only network SBML by inserting XML code. Attributes are written like Perl-variables into the annotated text. XML Tag-structures are enclosed into "XML_BEGIN" and "XML_END" marks.
2. Reconstruction of metabolic networks
Reconstruction of metabolic networks is the first step of rk_modelling and comprises collection and curation of biochemical reactions. The program design allows efficient use of existing knowledge in form of already published metabolic networks for the reconstruction of new metabolic networks. One advantage is that biochemical reaction can be directly copied to the user data. The resulting datasets can be modified: For example the biochemical equation and compartmentalization can be changed and Web-links, notes, remarks and variable declarations can be added.Context-menu
The items in the browsable trees have a context-menu (right-click) which give access to specific program functions.Cross-linking by ID
Datasets may have the same ID as a reaction in a loaded immutable network. In this case the dataset and the reaction are linked together. The dataset acts as an annotation of the reaction.Cross-linking by equivalence of the reaction equation
Reactions and datasets are also linked if they have the same biochemical equation. All reactions and datasets with equivalent biochemical equations are cross-linked. Reactions with a linked dataset are marked with a green (reaction exists) or red (reaction is not part of the model) traffic light sign. Unfortunately, different authorities such as KEGG, the Palsson group, ChEBI, HepatoNet and Reactome prefer different identifier systems for metabolites. Nevertheless, tables of equivalent metabolite identifiers can be provided such that reactions are recognized to be identical even if they are written with different identifiers. The equals relation for biochemical equations is important for the graphical user interface. Metannogen uses the graphical pathway maps from KEGG. In the pathway maps, reactions and metabolites can be highlighted by and quantitative data such as flux strength can be shown. Metannogen can be run without graphical user interface in a non-interactive manner to generate output files as part of a shell script.Program Features
The user interface follows conventions of typical graphical applications and implements advanced GUI concepts like browsable trees, , , rch and highlighting, -check and automatic Web links. Metannogen does not need to be installed because it can be directly started by clicking the Java-Web-start-button on the Metannogen web site. Using a central data store, several people can work simultaneously on the same project. To evaluate Metannogen, the demo database can be selected. The button to select the demo database is found in the start frame. The demo database is configured to accept simultaneous changes without password, whereas real projects should be password protected. Metannogen supports interprocess communication via socket connection to work in concert with other applications. is an XML based language for representing models of biological processes. It can be imported by many modelling programs.Central storage of datasets
A Metannogen dataset contains a biochemical equation and expert opinion edited by the curator. The datasets can either be stored in a local file on the PC or on a central HTTP server such that several investigators can work on the same data. When Metannogen is started with GUI the start dialog appears. The access data for a central data repository is entered into the top panel of the start dialog. In case the user wants to store the data locally, then the absolute file path is entered in the bottom part of the dialog.For using a central data repository, the user may create a repository on the www.bioinformatics.org-server. He will need to write down the access date which needs to be entered into the multi-line text-field. Alternatively, it is possible to store data on any other web server. This requires installation of a PERL-script. The following may be used as a template:
http://www.bioinformatics.org/strap/metannogen/metannogen.perl.txt.
 |
Server address: The multi-line text-field holds the URL of the PERL-script. The first valid URL is the master server. Optional, secondary URLs in text lines below may serve as Backup servers, as they receive the same data from Metannogen. The server can be tested with the Web-browser: Visiting this address in the Web-browser should show a file with all datasets. For example visit the demo server address:
http://www.bioinformatics.org/strap/cgi-bin/metannogenDemo.plLines that are commented out by a leading "#" are ignored.
Address of one single dataset The server address may optionally be followed in the same line by an expression denoting the URL for single datasets. The asterisk of this mask stands for the dataset ID. The URL of a single dataset is formed by replacing the asterisk by the ID. For example consider the URL mask of the demo server:
http://www.bioinformatics.org/strap/metannogen/demo_datasets/*.datasetReplace the asterisk by a dataset ID such as "R08689" and visit the resulting address in the Web-browser: This address allows Metannogen to observe the "Last-Modified" and "Date" attributes of the current dataset. If a dataset is uploaded by another investigator while it is modified, a warning message appears. The goal is to prevent data loss when datasets are modified simultaneously by different users.
Password: For password protected repositories, the server URL may contain a password in the form &passwd=xxxx. If a the password is not contained in the URL, the user can specify a password before uploading. If the password is not validated by the PERL script on the server, the user will not be allowed to change datasets.
Metannogen dataset
The datasets contain the data entered manually by the user. They have two different functions depending on whether Metannogen is used for Network reconstruction or whether it is used for SBML-file annotation. For Network reconstruction it contains all reactions of the reconstructed model. When annotating an SBML file, it is an annotation of that reaction in the SBML model, that has the same ID. A dataset contains a biochemical reaction written with metabolite identifiers.ID: Each dataset should have a unique identifier. If the ID is matching the ID of a reaction, then the dataset acts as an annotation of the reaction.
Creating datasets: An empty dataset can be created from the dataset-menu (Menu-bar>Datasets>
 ).
A panel with the dataset form is opened (see figure dataset-view).
It is also possible to use a reaction of one of the loaded networks as a template such that the
identifier field and the equation field will be filled.
The respective menu item is found in the (Right click) for reactions.
).
A panel with the dataset form is opened (see figure dataset-view).
It is also possible to use a reaction of one of the loaded networks as a template such that the
identifier field and the equation field will be filled.
The respective menu item is found in the (Right click) for reactions.
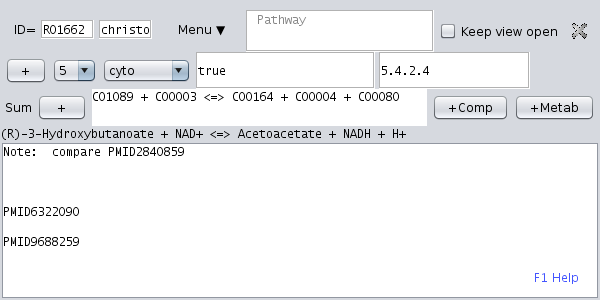 |
User interface: A dataset view is opened when the respective tree node is activated in the tree view or with the respective context menu. A dataset has several fields containing text. The different fields of the dataset are manipulated by appropriate graphical elements such as single and multi-line text fields, choice menus and toggle buttons.
 : The text-field for the chemical equation requires metabolite identifiers.
Below this text-field the equation is translated into a human readable biochemical equation using metabolite names.
Compounds can be typed directly into the text field either by name or ID. Typing of metabolite names is facilitated by .
Alternatively, compound names can be selected from a list
: The text-field for the chemical equation requires metabolite identifiers.
Below this text-field the equation is translated into a human readable biochemical equation using metabolite names.
Compounds can be typed directly into the text field either by name or ID. Typing of metabolite names is facilitated by .
Alternatively, compound names can be selected from a list  .
Transporters are equivalent to biochemical reactions but involve two or more different compartments.
Unlike enzymatic reaction where the compartment is set once for all
metabolites, each compound has its individual compartment written as
suffix.
For example "C00004@cyto" designates cytosolic NADH.
.
Transporters are equivalent to biochemical reactions but involve two or more different compartments.
Unlike enzymatic reaction where the compartment is set once for all
metabolites, each compound has its individual compartment written as
suffix.
For example "C00004@cyto" designates cytosolic NADH.
 /
/  : The activated state of this toggle indicates that the reaction is accepted as part of the network model and will be exported to SBML.
: The activated state of this toggle indicates that the reaction is accepted as part of the network model and will be exported to SBML. : Close the view of the dataset. The dataset is not lost.
: Close the view of the dataset. The dataset is not lost.- [x]Keep view open When this check-box is activated, the view remains open even when the views of other datasets are opened. This option is used to simultaneously view more than one dataset. When more than one view is opened, two alternative layout options are available and can be selected with a check-box: Vertical arrangement and pane.
 : This choice menu selects the compartment[s] for this reaction.
The list of all possible compartments can be changed by a command line switch.
: This choice menu selects the compartment[s] for this reaction.
The list of all possible compartments can be changed by a command line switch. : This choice menu allows to specify the confidence of the user that the given compartment is correct.
: This choice menu allows to specify the confidence of the user that the given compartment is correct.- The multi-row text area: This text-area contains free text.
Line-ends are internally saved as .
URLs and database x-refs act as hyper-links.
Pubmed and Uniprot x-refs are special because the abstract or protein file is automatically downloaded and displayed when
mouse is moved over the link.
This text area may also contain attributes for the reaction. These are written as a Perl-like variable declaration with a leading dollar sign. Example:$requires="low pH"
An attribute may be confined to one compartment. Example:$requires@lyso="low pH"
The text area provides standard functionality such as , , , auto links and text search and highlighting. A comprehensive overview of the functionality of the text area is displayed on pressing F1. The optional variable $HL can contain search strings that are highlighted in abstracts and PDF texts.$HL="sugar glucose mannose"
To highlight glucose green and mannose red type:$HL="sugar #00ff00 glucose #ff0000 mannose"
PUBMED links: Like all x-refs also Pubmed identifiers in the text area such as "PMID16020471" act as hyper-links. A prompt display of Pubmed abstracts (and Uniprot entries) is implemented using a cache: When the mouse pointer hovers over a Pubmed identifier, the abstract is downloaded. Once it is downloaded, it is always shown without delay when the mouse is over the Pubmed identifier and then displayed. The same applies to Uniprot identifiers. PDFs can be associated to Pubmed Identifiers using the context-menu (right-clicking the Pubmed-ID).
As text highlighting capabilities of document viewers like Evince or Acrobate are still limited, advanced highlighting features have been implemented for abstracts as well as for publication full texts within the Metannogen environment: The user can define a set of text patterns to be highlighted in the publication text (Menu-bar>Options>
 ).
These patterns apply to all datasets. Dataset specific catch words to
be highlighted can be defined by declaring a variable $HL=" pattern1
#00FF00 pattern2 ... " in the comment text field. Each token my be
preceded by a color written in hexadecimal HTML-syntax. For example
#00FF00 denotes the color green.
).
These patterns apply to all datasets. Dataset specific catch words to
be highlighted can be defined by declaring a variable $HL=" pattern1
#00FF00 pattern2 ... " in the comment text field. Each token my be
preceded by a color written in hexadecimal HTML-syntax. For example
#00FF00 denotes the color green.
Internal links: In the comment text of a dataset, references to other datasets can be included such that the referenced dataset is opened when the link is clicked. The links are typed in the form DATASET#datasetIdentifier. In addition, paragraphs in the comment text field can be referenced. This is useful, when two datasets share parts of the same text note. The paragraph must have at least a BEGIN#myLabelText anchor of the form BEGIN#myLabelText and may optionally be terminated with an end anchor like END#myLabelText. A tagged paragraph can be referenced. The corresponding references to this paragraph have the form #myLabelText or INCLUDE#myLabelText. In a similar way links of the form NETWORK#R00149 act as an hyper-link to all reactions (KEGG, Recon1, EHMN) with the given ID. Any metabolite identifier or any metabolite name has a context menu.
Additional GUI elements: It is possible to add additional GUI elements which correspond to specific columns in the Tab-separated Metannogen file format. These customizations must be performed by the project manager with sufficient programming skills. See: Menu-bar>Options>
 .
.
Additional additional dataset fields: The above approach requires programming skills. A much simplier way to structure the data is to use variable declarations in the comment text field. Example:
$myVariable="my text"It is possible to define an input form with variable declarations. A form may contains contains free text with embedded empty variable declarations. The curator can fill in the form by adding text to the empty declarations. The form is prepared with a text editor. No programming skills are required. The file path or URL of the form file is provided as a command line option.
Starting Metannogen - command line options
Metannogen requires Java version 1.5 or higher. It consists of a single -file metannogen.jar. Further data-files or program parts are automatically downloaded at runtime from the Web-server. Therefore the computer needs to be connected to the internet. The file metannogen.jar can be started in two different ways:-
Using the java-command:
java -Xmx200M -jar metannogen.jar [options]
The option -Xmx200M increases the maximum amount of memory allocated by the Java Virtual Machine to 200 MBytes. - Using (): The command javaws acts on the file metannogen.jnlp.
The JNLP file may be inspected with any text editor. A modified copy can be saved.
It contains the command line parameters and the the value for the maximum memory.
javaws http://www.charite.de/bioinf/strap/metannogen/metannogen.jnlp
Web proxy
In some institutions all Internet connections are going through a Web proxy (see ). In this case the proxy must be set correctly, otherwise downloading of files from the Web will fail. Detailed explanation is found in "Menu-bar>Options>Internet settings" or by clicking the button "Test Web Proxy" in the start frame of Metannogen.
Command line parameters of Metannogen
Metannogen takes a number of optional command line parameters. If Metannogen is started using the Java-Web-Start mechanism, the command line options are defined in the JNLP file. Some command line options are followed by one or more text files. Files can be given as a relative or absolute file path or in form of an URL.
General format of dictionaries:
For the syntax of most dictionaries two options exist:
-
Lines with Tab-separated entries:
If the dictionary file contains tabulator characters then
Metannogen assumes that each line contains a key-value pair such that both are separated by tabulator.
The first column in the tab-separated file contains the keys and the second column the values.
This can be altered with a suffix of the URL or file path. For example appending "(3,4)" to the url, the key will be read from the 3rd and the value from the 4th column (numbering starting with "1"). -
Lines with Space-separated entries:
However, if the file does not contain any tab, then Metannogen assumes that white space is the separator of keys and values.
With space as separator, lines with different keys but identical values might be combined to one line.
The advantage is, that the resulting file is more compact.
For example the two lines:
C00124 C00962 C01582 C00962can be written as one line (If the file does not contain any tab-character.)C01582 C00124 C00962
-
-networks List of files
The given SBML files are loaded into Metannogen and can be browsed offline. They form a repository of information that can be used for network reconstruction. Three widely used networks can be referenced by abbreviations, for all other networks the full URL or file pathe of the SBML file must be written.- "KEGG" is a symbol for the database,
- "RECON1" stands for the Human Metabolic Network of the Palsson group http://www.charite.de/bioinf/strap/metannogen/data/Palsson_human_2007_04_ascii.sbml.gz
- "EHMN" designates the Edinburgh human metabolic network.
- Networks within network databases can be given with the database ID followed by the network ID. Examples: JWS:Lambeth BIOMD0000000001
- -dictionaryOfSpecies List of text files containing identifier replacements
Example:... C00124 C00962 # D-Galactose C01582 C00962 # D-Galactose C00984 C00962 # D-Galactose C00159 C00936 # D-Mannose C02209 C00936 # D-Mannose ...Read: "Replace metabolite C00124" by "C00962". The value to be replaced is at the left and the replacement is at the rigth position. The identifiers are separated by white space. The same can be written in a more compact way:... C00124 C01582 C00984 C00962 # D-Galactose C00159 C02209 C00936 # D-Mannose ...The SBML output will contain C00936 even if C00159 or C02209 appear in the biochemical equation. - -dictionaryOfCompartments List of text files translating compartment names.
For some reactions the exact subcellular localization may be found in the literature, while for other reactions less specific information may be found. Metannogen supports subcellular localizations of individual reactions at level that is more specific than the final model. For example there may be evidence that a reaction takes place at a certain part of a golgi. This information may be recorded by the curator. But for metabolic network analysis distinction between cis- and transgoli is not suitable. Example:... cisGolgi golgi transGolgi golgi ...or shorter... cisGolgi transGolgi golgi ...The compartment names are separated by white space. The left compartment name is replaced by the right compartment name. In this example the SBML output will contain only "golgi" and not "cisGolgi" and "transGolgi". During data curation the compartments are recorded in more detail than needed. - -names List of text files for associating a human readable name to metabolite identifier
The two columns are separated by tab.... C05928\t10-Formyltetrahydrofolyl C05931\tN-Succinyl-L-glutamate ... - -replaceInEquation List of text files providing a dictionary for metabolite replacements.
This replacement is applied when the network model is generated from datasets. The replacement is performed before the dataset is parsed. It is possible to replace sets of species by single entries. For example the following line is valid:LEUKOTRIENE\tleua4 leub4 leuc4 leud4 leue4 leuf4Having this declaration, typing "LEUKOTRIENE" in the equation is semantically equivalant as typing "leua4 leub4 leuc4 leud4 leue4 leuf4". This notion might be useful in conjunction with brace expansion for example for transmembrane transport processes. Please note that in this example the first whitespce is a tabulator and the following white spaces are space-character. - -replaceWhileTyping List of text files providing a dictionary for metabolite replacements.
The replacement is applied in the text-field for the biochemical equation if the respective option is activated in the Menu-bar>Options. When biochemical equations are typed, the specified replacements will be applied automatically after the user has finished typing a metabolite. This is triggered by inserting a white space character after the identifier or by inserting longer text using Ctrl-V or the middle mouse button. In the example below C00022 is the KEGG identifier for Pyruvate and pyr my be a display name for pyruvate. If the user types "pyr" and hits the space bar, "pyr" would be replaced by "C00001".... H2O water C00001 pyr C00022 ...Loading networks like KEGG or RECON1 automatically activates replacement of metabolite names by identifiers. This replacement works independent of the option "-replaceWhileTyping". The identifiers and names are separated by white space. - -dictionaryOfSimilarSpecies List of text files associating a representative identifier to similar compound identifiers.
Example:... C01342 C00014 # Ammonia NH3 NH4+ C01353 C00288 C00011 # H2CO3 HCO3- CO2 ...Biochemical reactions are sometimes written in different ways as a question of taste: Either with NH3 or NH4+ or either with CO2 or HCO3-. As a consequence, Metannogen may fail to recognize that two biochemical equations denote the same catalytic process. This is avoided by providing a dictionary of similar species metabolites and by ignoring H+. Including NH3/NH4+ and HCO3-/CO2 into the dictionary, for additional 53 reactions in KEGG an identical counterpart is found in Recon1. The identifiers and names are separated by white space.
This dictionary does not affect network output. It is only used in the GUI to determine whether two reactions are identical. - -proton List of identifiers designating protons. When two reactions are checked for identity, protons are not regarded. To recognize that two reactions are equal if they differ only by one H+, the metabolite identifiers for H+ must be known. This is used for the GUI but does not affect the exported model.
- -datasetForms List of input masks for datasets. Input masks are free text containing empty variable declarations to be filled in by the curator. This allows to enter information in a structured form. All information written in variables are accessible with the API and can be used for the output file.
- -listOfCompartments File with a list of compartments.. This list is used for the compartment selector menu.
- -mapAffymetrixRid file or URL
Mappings of or identifiers to reaction identifiers. This dictionary is important for two features in Metannogen: Expression values provided in a -separated_value text file can be displayed graphically in the KEGG Pathway maps. Each text line of the data file is assigned to one or several KEGG reactions. If a text line contains a KEGG identifier then the assignment is straight forward, but if the lines contain other identifiers such as, Ensembl- or Affymetrix-identifiers the respective KEGG reaction is found with the use of the dictionary. The dictionary is also used for highlighting certain reactions in the graphical map using a list of KEGG identifiers or Affymetrix or Ensembl identifiers. A reaction is selected by ctrl-left-click the reaction box in the graphical map. It is unselected by shift-ctrl-left-click. The file as the following structure:... 208308_s_at \t R02740 204704_s_at 202022_at 200966_x_at \t R01070 R01068 ...In the left column are one or several or identifiers which denote a gene or an oligonucleotide, respectively. In the right column are one or several reaction identifiers. Please note, that a respective mapping is already automatically generated from the KEGG database for human and murine Ensembl identifiers via KEGG-orthologies but for other species the user must provide the mapping. - -customizeAddScriptByRegex File with List of shell scripts
Executing shell command. In general, customizable text can be changed with the option -customizeAddName.
Metannogen for network reconstruction - non-graphical mode
This chapter explains, how a network reconstructed with Metannogen can be exported using different file formats like SBML. This does not apply to the case that metannogen is used to annotate an SBML file. Though the file export function is accessible in the GUI (Menu-bar>File), the prefered way is to run Metannogen as a command line tool without GUI. This allows Metannogen to be included in shell scripts. Complex pipelines can be constructed to directly couple Metannogen with data analysis. The option "-datasets" and an option like "-toSBML1" are required. The other options are optional.- -datasets File with datasets
Loads the dataset files from the given file or URL. The URL usually points to a Perl or PHP script which concatenates the single text files of each dataset to one text files. Two possible applications:- When Metannogen is used from another program to access the data programmatically. In this case the other program invokes:
charite.christo.metannogen.Metannogen.main("-datasets","file.txt"); - To export the data in non-interactive mode. In this case it is used together with an option like -toSBML1 output-file
- When Metannogen is used from another program to access the data programmatically. In this case the other program invokes:
- -toModifiedSBML OutputFile
Example:java -jar metannogen.jar -datasets datasetFile -networks "http://www.ebi.ac.uk/biomodels-main/download?mid=BIOMD0000000001" -toModifiedSBML output.sbmlThe given file here BIOMD0000000001 is written to output.sbml. Annotations found in datasetFile are inserted into output.sbml. - -toSBML1 OutputFile
The customizable output script No 1 is used to generate output. If script No 1 was not modified by the user, then the default SBML output is used. This command line option requires, that the dataset source is specified with the -datasets option.
Different versions of export scripts: The user may also refer to Export_SBML2.java by using the option -toSBML2. Each export script Export_SBML[number].java can be modified at runtime using the GUI. Without restarting Metannogen, the modified export script can be run to export the file. The following options are analogous: -toSBML2, -toSBML3 ... or -toTXT2 or -toSQL2 or -toLIBSBML2. The latter requires installation of the program library libsbml (http://sbml.org/Software/libSBML). Detailed instructions are found in the source code of the export script and in the respective tutorial. - -attributes List of text files with variable declarations
The two columns are separated by tab. The first column contains the dataset identifier, the second column contains one or several variable declarations separated by white space. Content containing white space must be quoted with double quotes.... R00959\t$VAR1="variable one for dataset R00959 " $VAR2_cyto="This is only valid for cytosol" R00960\t$VAR1="variable one for Dataset R00960 " $VAR2_cyto="This is only valid for cytosol" ... - -annotationFormats List of text files with annotation formats
The text files describe how attributes like $IS_PART=" UNIPROT:P0A7B8 UNIPROT:HSLV_ECOLI " are translated into annotation texts like The two columns are separated by tab. The left column contains the attribute name and the right column the xml text. The files contains also the mapping of Metannogen database IDs to Miriam-urns. The string "REPLACE" will be replaced by the respective urn:ID. The file http://www.bioinformatics.org/strap/metannogen/data/annotationFormats.txt will always be loaded. - -useReactionAttributes List of attribute names for export.
Attributes contain additional information for elements (Also see http://www.w3schools.com/Xml/xml_attributes.asp). In SBML several standard attributes for reaction and species elements are well defined. Nevertheless, the possibility for additional (non-standard) attributes had been included in Metannogen for two reasons.- The set of standard attributes may be extended in future SBML versions.
- A computer program that takes SBML as input may accept information in form of non-standard attributes.
- -useSpeciesAttributes Equivalent to "-useReactionAttributes".
- -attributesOverride List of text files with variable declarations
Same as "-attributes" with the difference that the variables override the variables defined in the comment text of datasets. - -exportReactionIDs List of text files providing a set of reaction identifiers to be exported
R01271 R00301 HR05020.*Tokens containing asterisk are treated as ion. The compartment suffix like "@cyto" ist optional. If this option is given, only the specified reactions are included in the output. All other reactions are skipped. - -exportReactionIDs one file defining shell scripts which are applied to
selected texts and Object IDs
- -noExit
Metannogen will not terminate the Java-process. This option is only important when the main-method charite.christo.metannogen.Metannogen.main(String[]) is called from another program.
Specifying the command line options jnlp file: The command line options are specified in the jnlp file that is behind the Web-start button. This file need to be modified when other program options are required. It can be downloaded to the Desktop and edited with a text editor. When the file is clicked on the Desktop Metannogen is started.
Brace Expansion of the biochemical reaction
This paragraph applies to the case that the Metannogen is used for reconstruction of metabolic networks but not for the annotation of SBML files. Some enzymes and transport carriers are unspecific. To avoid creation of many similar datasets, can be used. Brace expansion allows to define several similar biochemical conversions within one single text string. If the reaction string entered by the curator contains curly braces then Metannogen applies Brace expansion to determine the set of reactions resulting from the dataset. The result are several biochemical reactions each having a different metabolite from the group enclosed in curly braces. The number of resulting equations is the number of metabolites within braces. If there are several groups of metabolites enclosed in braces then every i-th metabolite within each group belongs together. If the number of elements is not the same in all groups then an error message is written out. Example for transport reaction
{ Alanine Proline Glutamine }@cyto <=> { Alanine Proline Glutamine }@mito
is expanded to
Alanine@cyto <=> Alanine@mito Proline@cyto <=> Proline@mito Glutamine@cyto <=> Glutamine@mitoExample for transaminase reaction
{Alanine Aspartate} + Alpha-Ketoglutarate <=> { Pyruvate Oxalacetate} + Glutamate
is expanded to
Alanine + Alpha-Ketoglutarate <=> Pyruvate + Glutamate Aspartate + Alpha-Ketoglutarate <=> Oxalacetate + GlutamateIf the second parenthetical group is identical to the first one, then it can be abbreviated by "{}". This is usually the case for transport reactions. There must not be space between both braces:
{ Alanine Proline Glutamine }@cyto <=> {}@mito
Advanced: Double brace expansion: A second independent group of metabolites can be included in double curly {{ ... }} brackets. The number of resulting reactions corresponds to the ct of the two sets.
Text-Pane short-cuts
The multi-line text-pane is derived from the standard Java text-pane and provides additional functionality. Shortcuts with the CTRL key: The control key is locateted at the left on the keyboard is labeled "Control", "Ctrl" or "Strg", IKIde:Steuerung_(Taste). The text-pane receives the key-strokes only if it has the input focus. Click to set focus.- X Cut Text and copy
- C Copy text.
- V Paste text.
- F Find text pattern. Visit http://www.bioinformatics.org/straputils/ for a complete description of search and highlighting options.
- L Toggle line numbers. Or toggle truncate/fold lines
- + Enlarge font.
- - Smaller font.
- * Toggle text quality (see ).
- T Toggle frame is always floating on top. Indicated in frame title.
- P If it is a HTML document a printable html document will be opened in the Web-Browser.
- ESC Disable links until the mouse exits and re-enters.
- F1 Help
- F6 Settings: Specify Web-Browser, File-Browser and Standard URLs
- F7 Spell check
- F10 Toggle
- ALT-slash or ctrl-ENTER and sometimes also Tab . File path completion if the word at the cursor looks like a file path. Also Environment variables are completed.
- Ctrl-Z
- Ctrl-Y
Hyperlinks
In most text views URLs like http://www.google.de or database references like 12166070 or PUBMED{fructose glucose} or EC classes like "1.2.2.1" can be clicked to open the selected item in the browser or document viewer. To specify another Web-browser hold the CTRL-key while clicking. The list of database links can be customized: ,
,
 ,
,  .
.
Context Menus
The context menu is opened by right-clicking a word. Pubmed links like PMID2840859 have a specific context menu.Auto-word-completion
available for certain text views. Typing the beginning of word and hitting the Tab-key or alt-/ completes the word. In case of ambiguity, the desired word may come up by pressing the key several times. nullContext menus and Balloon text
As the mouse is moved over an identifier in an editable or read only text-field, the name of the respective object is shown. For example moving the mouse over the metabolite identifier "C00984" brings up a with "D-Galactose". Metannogen associates "C00984" with the "D-Galactose" if KEGG is loaded. The same happens if the mouse is moved over "gal" if a Palsson network has been loaded. Names can not only be found in loaded networks, but may also be provided by a file given after the command line option "-names".Identifiers or names of metabolites, reactions and datasets have a . This context menu allows to locate the object in the object hierarchy of the network and to highlight it in the graphical pathway map. Related reactions and datasets can be opened via the context menu.
Browsable Tree
Several browsable trees are available in different tabs of the tabbed pane:- Tabs for networks like KEGG and the Recon1
- Tab for objects matching a keyword search
- Tab for clicked objects
- Tab for datasets
- Tab for datasets ordered by pathway
Multiple selection of tree nodes follows the conventions of Windows and X-Windows and requires the Shift key and the Ctrl key.
Importing biological Networks
Metannogen allows to load and browse finished networks like Kegg or Recon1. The immutable network data is shown as a hierarchical tree with expandable nodes: Pathway, reaction, EC-class, metabolites.Datasets
The datasets are listed in two trees located in adjacent tabs:- Datasets: Datasets are in flat lists: Several branches are available:
 : This branch contains all datasets edited and modified by the user.
: This branch contains all datasets edited and modified by the user. : This branch contains all datasets that have been deleted during the current session.
Deleted datasets can be rescued with the menu-item "Duplicate focused dataset" in the dataset-menu or by clicking the
: This branch contains all datasets that have been deleted during the current session.
Deleted datasets can be rescued with the menu-item "Duplicate focused dataset" in the dataset-menu or by clicking the  button.
All deleted datasets are finally lost after the session.
button.
All deleted datasets are finally lost after the session. : This branch contains all datasets that are not assigned to a reaction because the ID does not match to any reaction.
: This branch contains all datasets that are not assigned to a reaction because the ID does not match to any reaction. : This branch contains all datasets with more that one compartment.
: This branch contains all datasets with more that one compartment.
- By PW: Datasets are ordered by pathway. Only those datasets are included where a pathway is defined in the Pathway-text-field.
 |
Bugs: Sometimes the tree is not displaying any more. Workaround: "Redraw"-button.
Graphical KEGG pathway maps
The KEGG database provides graphical representations for several classical pathways. These pathway maps often resemble those in text books. They can be viewed in Metannogen by opening the context menu. The pathway maps are interactive i.e. the reactions and metabolites have a balloon message and a context menu. It is possible to view quantitative data of reactions on the maps.Export Data
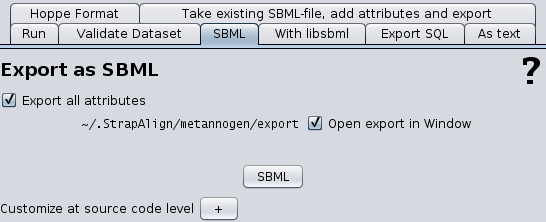
The export-Dialog is opened from the file-menu. It allows- to generate a new SBML file from a given network SBML file by inserting annotations. (Only applicable if Metannogen is used to annotate an SBML file.)
- to export the network reconstructed from scratch with Metannogen. (Only if Metannogen is used to reconstruct a metabolic network.)
- to analyze/process all data by own scripts
- to validate dataset during input
 |
- charite.christo.metannogen.Reaction contains the reactants and products, the compartments and optional attributes.
- charite.christo.metannogen.ReactionCollection contain arrays of Reaction and Species objects
- charite.christo.metannogen.Species contain the name and attributes of reactants or products. The Species class is not always required because the Reaction class can return metabolites also as plain String objects.
- charite.christo.metannogen.Metannogen gives access to the Networks. Network files are loaded according to the -networks=file-list command line option. It also provides a function to create a network from all datasets. Metannogen has the main(String[]) method which is called when metannogen.jar is started.
Executing shell command
This Dialog allows to apply shell commands to selected text regions and object IDs. It provides one way of linking other programs. The list of shell scripts can be customizable. The user can chose the shell script to be applied to a certain text-string from a browsable tree. This hierarchical tree contains the menu and sub-menus and shell-scripts and Web addresses. |
The list of shell-scripts is customizable at run-time. Alternatively, the program option "-customizeAddScriptByRegex" can be used to specify a file with shell-scripts. Example:
demo\Example google%09.*%09http://www.google.com/search?q=* demo\Example: Matches starting with M%09^M.*$%09 echo starts with letter M *Each line associates a shell script (3rd column) to regular expression (2nd column). The asterisk is replaced by the text. The shell script is presented to the user as a menu item. The menu item path is given in the 1st column with back-slash being the separator. If the value in the 3rd column starts with http://, then it is not taken as a shell script but as a Web address.