What is Co-Evolution?
Imagine you are studying all the doors in a large building. These doors all share some features in common - they are likely all recentangular, with hinges.
These are 'conserved' features, that are clearly important for the basic function shared by all doors. Other features, such as size, colors, and trim, are not shared. One might (correctly) conclude that these features are not important for door function.
There is, however, another feature that might exist on most doors - the lock. This also may be variable, or not highly conserved. Thus, at first glance this would appear to be an unimportant feature.
However, if we simultaneously observe all the keys that exist for this building, we will notice a co-incidence; the locks and the keys co-vary between doors. This suggest that both are important for function, and moreover, that they form a specific interaction when carrying out this function.
We can carry this analogy of co-variance to molecular sequences of proteins and nucleic acids. While there is no builder to vary 'doors', natural selection does the same job, and has produced some features of molecules that co-evolve. This co-evolution can indicate that there is a sequence specific, functionally important interaction between two residues. These residues can be within the same molecule (intramolecular) or across two different molecules (intermolecular). Most importantly, we can potentially detect co-evolution simply by observing sequences, for which we will use bioinformatics tools.
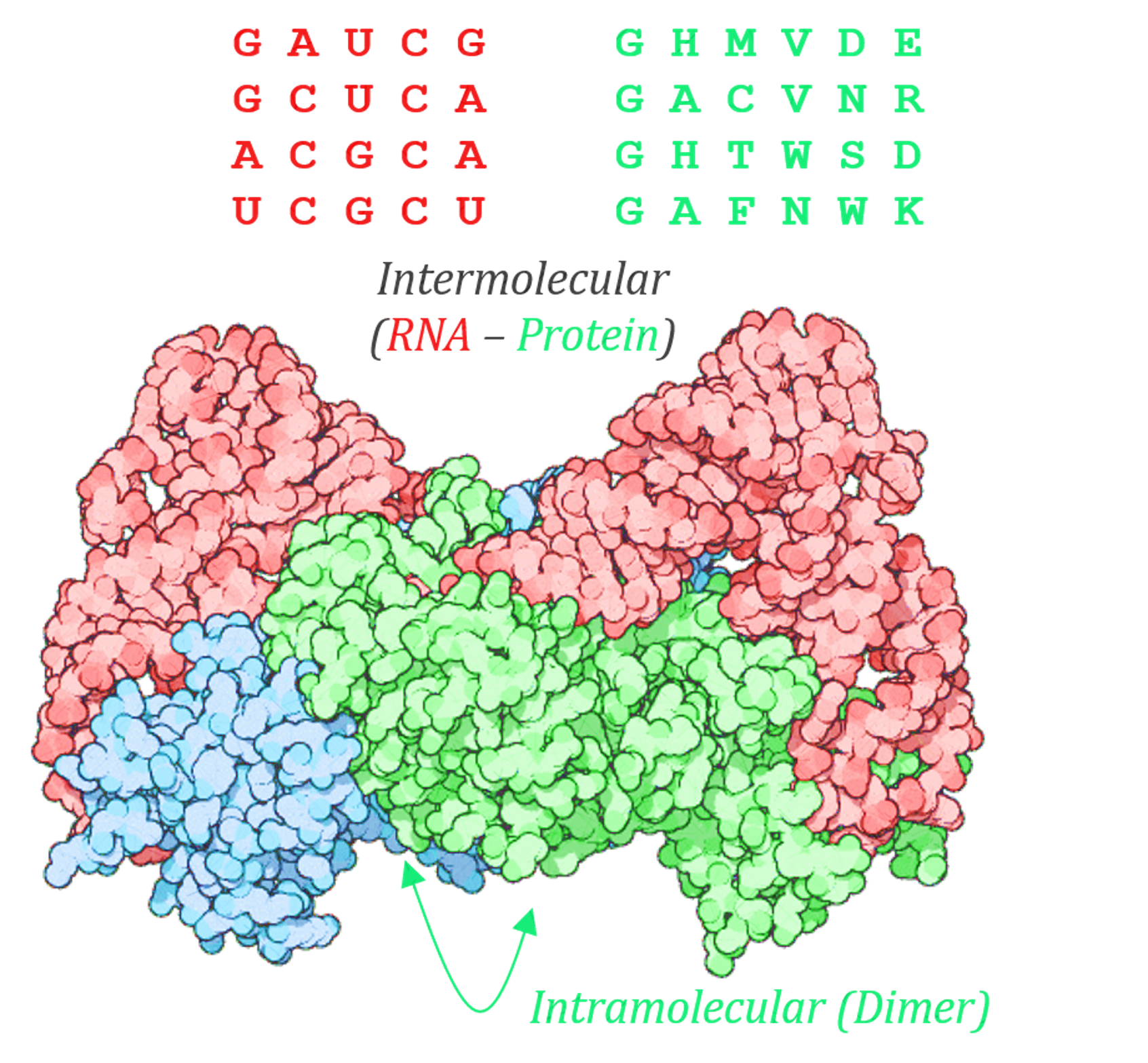
Molecular co-evolution between a protein (green) and an RNA (red) is demonstrated in the example above. In this hypothetical example, there is both intramolecular co-evolution (caused due to an interaction at a dimerization interface of the protein, typified by columns or residues 2 and 6 in the sequence shown) and intermolecular co-evolution (Typified here by an interaction or co-variance between the 3rd residue in the RNA and the 4th residue in the protein).
Why is it important?
Interactions between residues and subunits of molecules such as proteins and nucleic acids help govern many important cellular behaviors. Thus, to understand these behaviors, and to gain the insight necessary to modify and engineer them, we must ultimately understand the aforementioned molecular mechanisms and interactions. Highly conserved residues are often important for molecular function, but these are mostly invariant among organisms and cannot be easily reconfigured. However, residues that appear poorly conserved may still be critical for molecule function, if the evolutionary changes that occur in that molecule are compensated by simulatenous changes in the salient interacting molecule (or, for molecule structure, within the same molecule). Two residues that display this pattern are said to co-evolve.
Identification of co-evolving residue pairs is significant and worthwhile. These residue pairs would be predicted to form functionally-important, sequence specific interactions (Of course, the bioinformatic predictions must be validated by experiment). Unlike highly conserved residues, these pairs can be reconfigured to generate new interactions or new cellular behaviors, or to develop orthogonal sets of interactions in an organism.
What is this site, exactly?
This site has a collection of software that can be useful for anybody that is attempting to assess molecular co-evolution. These tools, together with external ones, can create a pipeline for sequence retrivial, processing, and analysis.

These original software tools are available by following the links below, or visiting the download index. Most of these tools are available in a .zip package, which contains a .jar executable (requires Java be installed on your machine), readme files and some example input and output.
I recommend using the downloadable versions of these files for working offline - for some of them, the processing time makes it easier to run on your local device. For the simplier programs, javascript or PHP webserver versions may be available now or in the near future.
Site Last Updated on January 20th, 2023. Check back often for updates!
A special thanks to Christopher Camenares, who provided useful coding advice and example code.

Sequence Retrival Tools: A variety of simple software tools and tips for programmatically retrieving large sets of sequences.More...

Sequence Name Filter: Eliminates unwanted sequences from a collection based on their name.More...

Combine Fasta: Merges two or more FASTA files together, eliminating duplicates within and across the files.More...

Taxonomy Filter: Starting with two sequence collections, unsures that only sequences which arise from species represented in both collections are kept.More...

MSA Gap Remover: Given a reference sequence and an MSA, remove all positions that correspond to ref. sequence gaps.More...

MSA Concatanate: Fuses the MSA of two distinct molecules into one. Useful for preppings sequences for certain software tools (i.e. MISTIC2).More...
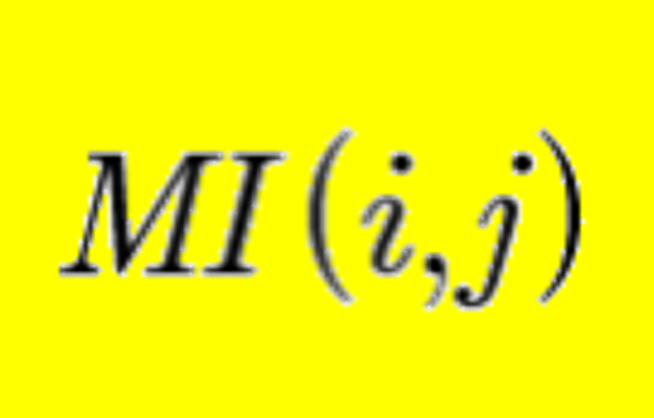
miBIO: Calculate Co-evolution within one molecule or across two different molecules.More...
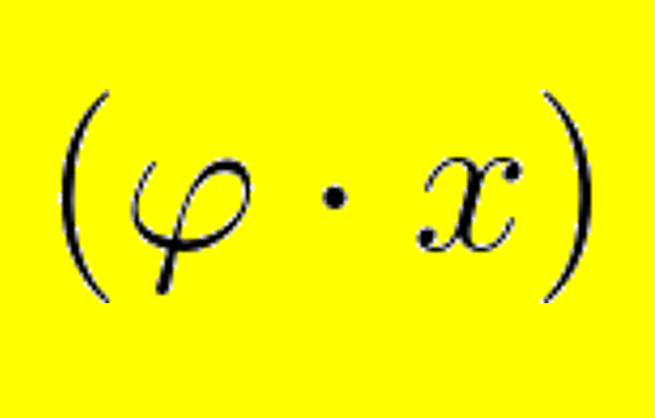
aCeS: Simulate co-evolution within and between several molecules. Useful for testing hypothesis or benchmarking software.More...



