- Chrome browser: Bringing new tabs to the foreground seems to be the default in recent versions of Chrome. If not, you can install the free extension Tab Activate.
- Safari browser: Click Safari in the menu bar at the top of your screen. Select Preferences, Tabs, and check When a new tab opens make it active.
- Edge browser: Bringing new tabs to the foreground seems to be the default. If you have more information, please share with the
- Opera browser: Bringing new tabs to the foreground seems to be the default. If you have more information, please share with the
- Firefox browser: Go to Preferences or Settings, then General, Tabs, and check When you open a new tab, switch to it immediately.
|
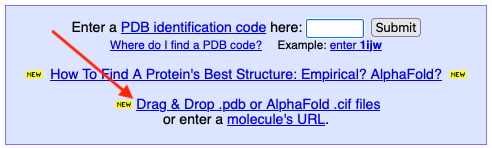
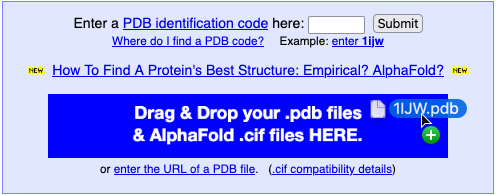
Clicking Drag & Drop
opens a blue landing pad →
Multiple files can be dropped on the same landing pad. Each will open a separate FirstGlance session. |
|
| An "empty basket" indicating the location of one or more missing residues. |
|
 Introduction panel in FirstGlance:
The Introduction fills the Lower Left Panel
at the beginning of a session. Later, you can get back to the Introduction by clicking
"Return to Introduction" at the bottom of the Lower Left Panel. See also Videos.
Introduction panel in FirstGlance:
The Introduction fills the Lower Left Panel
at the beginning of a session. Later, you can get back to the Introduction by clicking
"Return to Introduction" at the bottom of the Lower Left Panel. See also Videos.
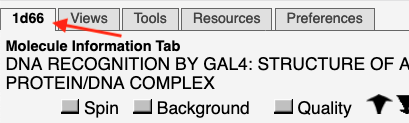 Molecule Information Tab: This tab is labeled with the PDB code unless the model is not an entry in the Protein Data Bank, or has been modified.
Molecule Information Tab: This tab is labeled with the PDB code unless the model is not an entry in the Protein Data Bank, or has been modified.
-
To reset a ConSurf result:
If you are in ConSurf View (stated at the upper left), click the button Reset View.
If you are in the FirstGlance controls (tabs at the upper left), click on ConSurf Controls (middle left), and after you get the ConSurf View (stated at the upper left), click on the button Reset View.

|

|
|
New default "Shiny" rendering. |
Older rendering, now a preference option. |
 Simplified Mode of FirstGlance: FirstGlance is initially in a simplified mode in which
fewer details are shown. Optionally, more details can be displayed (see
snapshot at right). Whichever mode you choose will be remembered between sessions
(provided you use the same browser and computer).
Please see more information.
Simplified Mode of FirstGlance: FirstGlance is initially in a simplified mode in which
fewer details are shown. Optionally, more details can be displayed (see
snapshot at right). Whichever mode you choose will be remembered between sessions
(provided you use the same browser and computer).
Please see more information.
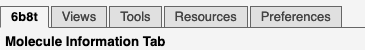 Tabs at the upper left of FirstGlance select different control panels.
See snapshot at right.
Tabs at the upper left of FirstGlance select different control panels.
See snapshot at right.
 Uploading
a PDB file from your computer:
The former uploading mechanism has been replaced by
Drag & Drop.
Uploading
a PDB file from your computer:
The former uploading mechanism has been replaced by
Drag & Drop.