![]()
Tutorials
How to load data into Assemble2?
You have: Assemble2 installed and configured.
You want: play with it!!
In Assemble2, a working session is based on a RNA secondary structure. Assemble2 allows you to get one from different kind of input data:
- a single RNA sequence,
- a set of RNA sequences,
- a secondary structure,
- a tertiary structure,
- an annotated genomic sequence.
-
FASTA files (File -> Load... -> RNA Molecule(s)): a FASTA file can contain one or several molecules. If several are available, you will have to choose your favorite one. Assemble2 will compute its folding landscape using CONTRAfold, RNAfold and RNAsubopt.
If several sequences are described in the FASTA file, Assemble2 will compute a structural alignment and infer a secondary structure for your favorite sequence (thanks to the algorithm mlocarna). As for all Sankoff-style algorithms, mlocarna can be very costly and time-consuming if you have numerous and/or large sequences. You have to note that all the gap characters will be ignored from a FASTA file.
-
Clustal files (File -> Load... -> RNA Alignment): Assemble2 can handle the classical format without any structural information or an extension of this format supplemented with a structural information. Without any structural information, Assemble2 will use the algorithm RNAalifold to predict a consensus secondary structure based on the alignment described. Then, it will infer a secondary structure for your favorite sequence.
The alignment will be displayed in the panel "Structural Alignment" and all the sequences will be listed in the panel "Aligned RNAs". If the Clustal file contains a consensus bracket notation at its end (see screenshot below), Assemble2 will use it to directly infer a secondary structure for your favorite sequence.
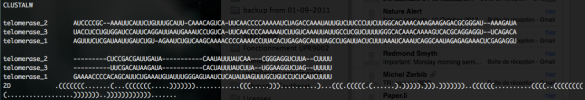
- Stockholm files (File -> Load... -> RNA Alignment): same behavior as with a Clustal file supplemented with a structural information.
- Vienna files (File -> Load... -> RNA Secondary Structure): this format is also known as "dot-bracket notation". Organized like a FASTA format, each biological sequence is linked to a bracket notation describing its secondary structure. Assemble2 will display the secondary structure described with the bracket notation linked to your favorite sequence. As with FASTA files, gaps are ignored.
- BPSEQ or CT files (File -> Load... -> RNA Secondary Structure): these formats describe a single RNA secondary structure.
- PDB files (File -> Load... -> RNA Tertiary Structure): Assemble2 will use the algorithm RNAVIEW to annotate the 3D structure. The RNA secondary structure inferred from the 3D information will be displayed in the main panel and the 3D structure will be displayed in Chimera.

Once the secondary structure for your favorite sequence loaded and displayed, Assemble2 provides your other ways to create secondary structures. You can:
- modify interactively the secondary structure displayed by adding/removing helices and tertiary interactions (see "how to edit an RNA secondary structure?" for details)
- modify the consensus bracket notation in the panel "Structural alignment" and infer a new secondary structure (see "how to construct and manipulate a structural alignment?" for details).

How to load genomic annotations?
Using "File -> Load... -> Genomic Annotations)", you can load genomic annotations from a GFF3/Genbank file, or directly from the Genbank database. If you load a GFF3 file, Assemble2 will ask you to choose a FASTA file storing the genomic sequences. This will open a lateral panel named "Genomic Annotations"
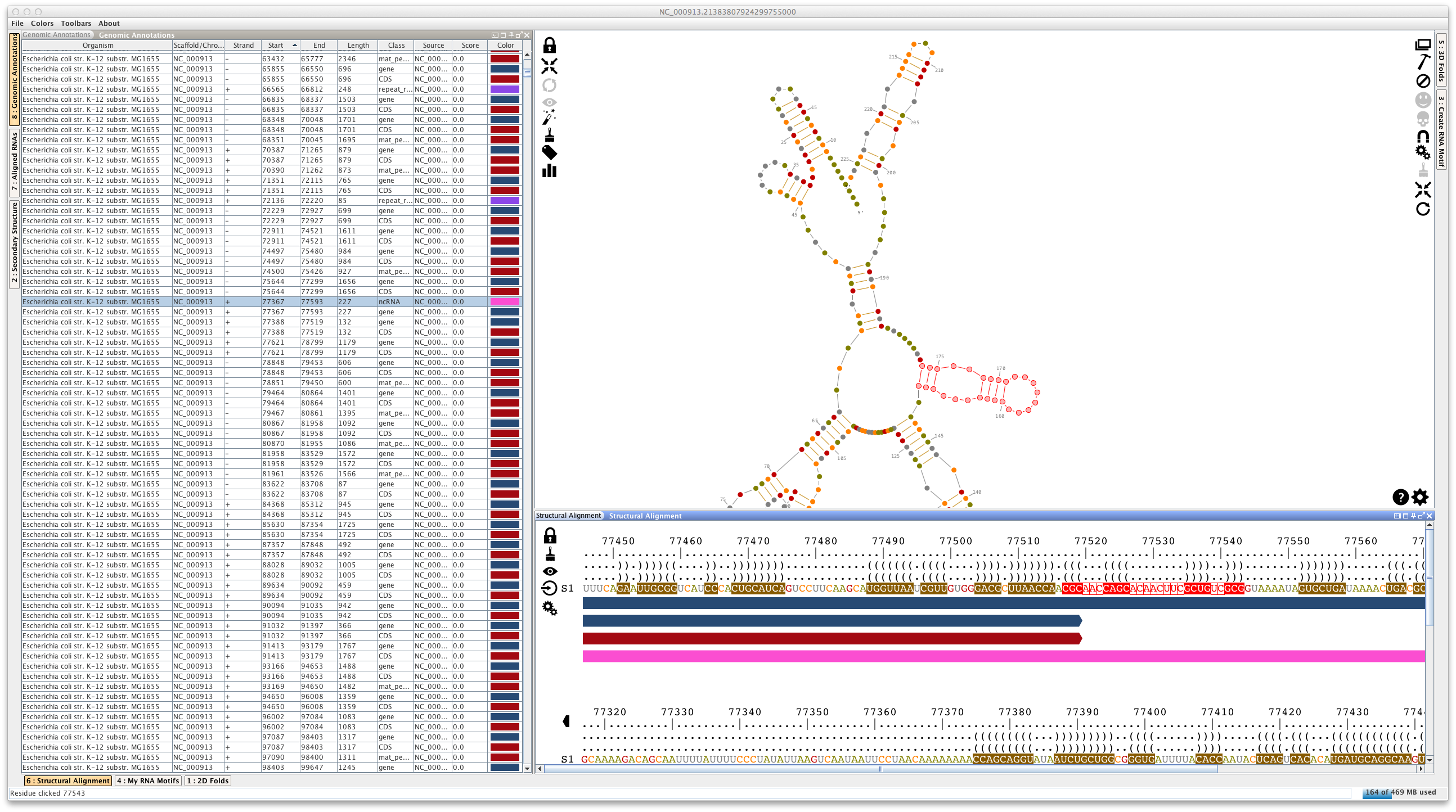
If you right-click on a genomic annotation, Assemble2 will compute its folding landscape and display it in the lateral panel "2D folds". Be careful of the size of the genomic annotation, otherwise the computation could take a while.