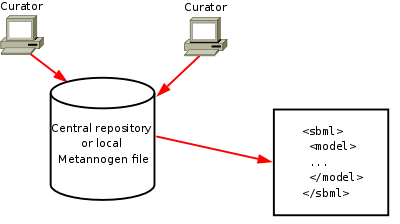
Metabolic network annotation
Metannogen
With this Java software you can (I) browse and annotate existing
biological networks and (II) reconstruct biological networks.
Please cite the paper in Bioinformatics Abstract PDF
Annotating existing networks

|
-
Free text
- Source of information (Database or literature cross-references)
- Private notes, remarks, to-do's
- Questions to colleagues
- Database searches: Google, Pubmed, Blast
- Predictions: Sub-cellular localizations
-
Controlled vocabulary. Syntax like string variable declaration in programming language BASIC.
Customizable input masks may be defined.
- Simple XML Attributes
- Rdf Annotations
- Highlighting rules
- Evidence level
$IS_DESCRIBED_BY="PUBMED:12345 UNIPROT:P0A7B8"
gives rise to the
following annotation of a reaction inside the SBML document:
<annotation>
<rdf:RDF
xmlns:bqbiol="http://biomodels.net/biology-qualifiers/"
xmlns:bqmodel="http://biomodels.net/model-qualifiers/">
<rdf:Description rdf:is="#metaid_test1">
<bqbiol:isDescribedBy>>
<rdf:Bag>
<rdf:li rdf:resource="urn:miriam:pubmed:12345"/>
<rdf:li rdf:resource="urn:miriam:uniprot:P0A7B8"/>
</rdf:Bag>
</bqbiol:is>
</rdf:Description>
</rdf:RDF>
</annotation>
The default rules for the text conversion are taken from the file
http//:.../annotationFormats.txt.
Further files with rules can be added with the program option "-annotationFormats".
Those annotations that can be generated automatically, are fed into Metannogen via line oriented tab separated files rather than typed manually.
Metannogen can be used as a plugin for graphical network design tools, providing advanced capabilities for annotating reactions. The communication between the App and Metannogen works simply by network sockets and is language independent. See Controlling Metannogen through other application and Controlling other application through Metannogen for technical details.
Reconstruction of metabolic networks
|
Start demo
- Click demo without Kegg
- Or demo including Kegg
(Very long download time at 1st start)
- Find the expandable tree for the network components of Recon1 and Kegg.
- Add annotations for the reactions of KEGG or RECON1 using the context menu (right click).
- Use the export methods in the "File" menu to create SBML files.
For own projects, the program options need to be customized as described in Starting Metannogen (See Overview program options).
Features of Metannogen
Text views
|
Data Management/Export
Kegg Pathway maps
|
- Metannogen can not only be used to reconstruct networks but also to annotate existing networks
- Implementation of the Miriam standard
- Several new editing aids like tab-key word completion, spell check
- Self defined shell scripts and web services for text words and text selections
- In addition to Web services where the parameters are encoded in the URL, also web services where the parameters are in the HTTP header (so-called POST )
- Find&Replace in many datasets/annotations
- Visualization of numeric data such as gene expression in graphical KEGG maps
- Improved responsiveness and reduced memory consumption
- Ready for inter-operation with other systems biology software
- Fixing problems of browsable trees
Availability:
GNU-License. Source code included in jar-file. Free of charge. Tested on Linux, Windows-XP (SP2) and OpenSolaris, MacOSX.Related projects
|
|
|
Bug reports and suggestions to
| @ | christophgil |  |
googlemail | . | com |
