Click to start Strap
Author: Christoph Gille
Institut für Biochemie, Charité Berlin
| @ | christophgil |  |
googlemail | . | com |
Interactive
Structure based
Sequences Alignment Program
Strap
The free computer program Strap aligns proteins by sequence and 3D-structure und Windows, Macintosh, Linux and Unix. Run the demo showing 6 sequences of the proteasome within a Strap session.
Secure Strap session
Strap requires Java. In all versions of Strap, modifying file operations are strictly checked to prevent damage of data on the computer. The Strap "Lite" version used for above demo has additional restrictictions to improve security: It does not have permission to run external programs nor to load further Java code.
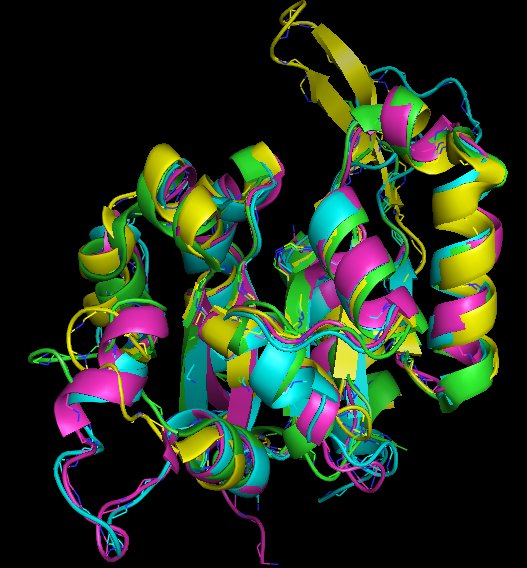
After starting the demo you will see 6 amino acid sequences. For three of them the 3D-structure is known. Those are well superimposed in 3D even though the similarity is hardly recognized at sequence level. Some important positions are marked in the sequence. These underlinings might be dragged into the 3D-view.
Strap supports the simultaneous analysis of hundreds of proteins and integrates amino acid sequence, secondary structure, 3D-structure and genomic- and mRNA-sequence and residue annotation.
 |
 |
Getting started - run demo
Click to run demo An alignment of orthologous and paralogous sequences of the core of the proteasome is shown. Each alignment row contains the amino acid sequence and the row header with the sequence name. The row headers have a context menu (right click) and can be moved/copied with the mouse (so-called ).Important sequence positions are highlighted after some time. A detailed balloon message appears when the mouse pointer is over the underlining. The information is freshly downloaded from various servers in the world and will be up-to-date which causes a delay of a few moments. They have a context menu (right click), they can be selected by Ctrl+click and can be copied by .
The 3D-structures are not directly available. This is because only sequence information is primarily present. Nevertheless, homologous 3D-models can be inferred using the tool-button "3D". This automated search for homologous structures is based on an up-to-date copy of the structure database. After association of 3D-coordinates from an homologous entry from the protein structure database, the structure can be viewed.
For those who are rather careful with unknown software:
If you do not yet trust Strap completely, run the light version with restricted
permissions - hence higher security.
Strap will not permit native code to be executed and will not load further java code.
Also see "Security" in the navigation panel.
Run light version
Web-Start
Java is needed. Click the "Launch"-button in the navigation panel which links the file strap.jnlp.Depending on the configuration of the Web-Browser, it may not work: In case of problems.
OutOfMemoryError
By default, Strap must not use more than 250 MByte. Therefore, Strap may throw an OutOfMemoryError.
Here you can start Strap with more memory.
OutOfMemoryError often occurs with the 3D-superposition
method CE for very large proteins. Solution: The method
TM_align requires less memory. The superposition method can
be changed with the context menu of the align-tool-button
(right click) or in the Superposition menu of the 3D-view.
The amount of free memory can be monitored in Strap in the left side pane which need to be opened with the mouse first.
Also see What_is_heap_size_in_Java
The amount of free memory can be monitored in Strap in the left side pane which need to be opened with the mouse first.
Also see What_is_heap_size_in_Java
Manual Installation and Deinstallation
: strap.jar (3.3 MByte)
Install manually if Web-start failed:
Download strap.jar (3 MByte) and double click or type the command line: java -jar strap.jar
Uninstall: Strap does not change the system. It does not modify global configuration files . All files reside in .
Previous program versions are found in the Archive or Archive before 2010
Uninstall: Strap does not change the system. It does not modify global configuration files . All files reside in .
Previous program versions are found in the Archive or Archive before 2010
 |
Documentation
- Manual (HTML)
- Unix style man page (HTML)
- Tutorials are found in [menu-bar > Tutorials] in Strap
-
Loading and saving of proteins is very important. The following three movies demonstrate, how
is used for proteins in Strap.
- Loading proteins from the file system (0.1 MByte)
- Loading proteins from Web resources - Example Uniprot (1 MByte)
- Loading proteins from Web resources - Example PDB (0.2 MByte)
- Copying proteins to the file system (0.1 MByte)
- Context menus (0.1 MByte)
- Loading proteins from the file system (0.1 MByte)
- With Strap you can handle 3D-structures. This is demonstrated in the following flash movies
Main features of Strap
- Computation of sequence alignments (Align-m, Amap, , Dialign, Dialign-T, JAligner, Kalign, Mafft, Muscle NeedlemanWunsch, ProbCons, T_coffee)
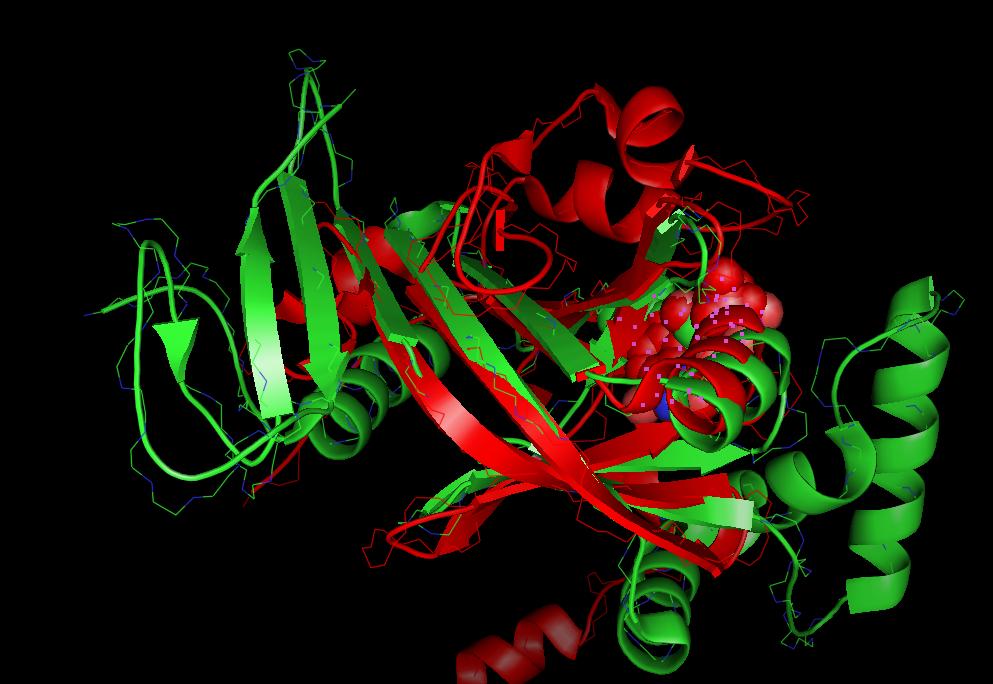
- Computation of 3D-superpositions (Example output) and structure based multiple alignments of proteins with TM-align or CE or Gangsta+ 3D-superposition, . Combined sequence-structure alignments.
- 3D-visualization using or Pymol or . In preparation: Highlighting mutations, SNPs, exon-intron boundaries or regions with high sequence conservation in 3D. Instant cross-selection between 3D objects, sequences and alignments
- Blast search including "Blast Alert"
- Prediction:Transmembrane helices, Secondary Structures and Coiled-Coil
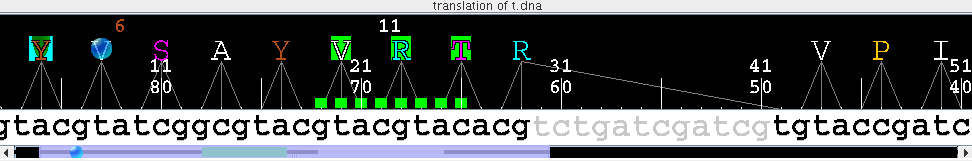
- Translation of nucleotide sequences to amino acid sequences. Output of amino acid alignments as nucleotide alignments.
- Protein file formats: , PDB, , MSF, Stockholm, , Nexus, HSSP , EMBL
- Phylogenetic trees with ATV, and GeneBee
- Residue annotations and nucleotide annotations
- Highlighting regular expressions (i.e. Prosite-Patterns) using and sequence features from BioDAS servers
- Aiding tricky sequence alignments due to low sequence similarity by:
- 3D-superposition of C-alpha atoms using and to highlight spatial equivalent residues
- Dotplot
- Intermediate Sequences
- Mixed sequence alignments and protein structure alignments
 |
Alignment Export
The sequence alignment can be exported in various formats:- ClustalW, MSF, HSSP, multiple Fasta
- ClustalW wih nucleotide triplets: Example
- HTML
- MS-Word, OpenOffice
- PDF: Example Conservation, Hydropathy, Fingerprint-overview.
- PDF with bar charts: Example Color encoding and Bar chart. See TeXshade.
Strap in Web-pages
 |
- Web-variables: A limited amount of information can be passed as web-variables in the address.
- Script commands: For complex tasks the scripting language can be used in the web page.
Strap is used as a viewer by:
The interaction of web pages and Strap is demonstrated in the
animation
(1.1 MByte).
Strap with reduced
functionality ( invisible menu-bar) serves as a simple web-viewer for
proteins. Static web links are clicked by the user to load
proteins. In the video four pdb files are loaded and structurally
aligned. The files are saved on the desktop by and
the alignment is shown in the web-Browser and in MS-Word.
(1.1 MByte).
Strap with reduced
functionality ( invisible menu-bar) serves as a simple web-viewer for
proteins. Static web links are clicked by the user to load
proteins. In the video four pdb files are loaded and structurally
aligned. The files are saved on the desktop by and
the alignment is shown in the web-Browser and in MS-Word.
eLearning
Strap has been used for blended eLearning in various modelling courses in the world. It will be used as an eLearning tool for students of human medicine. First results can be seen here: eLearningNews
- Nov 2005: BTI contact. Improving GUI
- Jan 2008: interface added
- Jul 2008: Serialization of alignments in Web-links
- Mar 2009: BioDAS servers for Sequence features
- Mar 2009: New 3D-Superposition Program: Gangsta+
- Jan 2010: added
- Mai 2010: Creation of a lite version. Tighter integration into Metannogen
- July 2011: Integrated
- August 2011: Integrated
- March 2011: Added to blog
- January 2012: Adding more 3D-alignment algorithms
- June 2012: Improving HTML/MS-Word export
- September 2012: Adding support for SQL based cache
Bug reports and suggestions to
| @ | christophgil | |
googlemail | . | com |
If a previously working feature is broken then use a Previous version and report the bug.

{kind=link}